
1.jpeg)
Introduction
History
In 1908 George Coats1 original description of the disease was based primarily on histopathologic examination of enucleated eyes and identified retinal vascular aneurysms, arteriovenous malformations, intra- and subretinal hemorrhages, and exudates. Coats categorized eyes with these characteristic morphologic findings into three groups: Group I demonstrated massive subretinal exudate alone, group II consisted of eyes with massive subretinal exudate, intra and subretinal hemorrhage, and retinal vascular dilatations, and group III included eyes with subretinal exudate and retinal arteriovenous malformations. Von Hippel2 later identified group III as a separate entity, angiomatosis retinae, which led to the exclusion of this group from the spectrum of Coats disease.
In 1912, Theodor Leber3 described a disorder with similar retinal vascular abnormalities to Coats disease, but without massive subretinal exudate, hemorrhage, and serous retinal detachment. This became known as Leber multiple military aneurysms, and Leber concluded that this entity was probably an earlier or less severe form of the disease previously described by Coats. In 1956 Algernon Reese4 observed patients with the clinical appearance described by Leber who eventually progressed to a Coats-like clinical picture, and further reinforced the notion that the two entities were part of the same clinical spectrum.
Coats disease can be defined as idiopathic congenital retinal telangiectasia with intraretinal and /or subretinal exudation and without appreciable vitreoretinal traction.
Epidemiology
Demography
Although Coats disease can be recognized at any age, most patients are diagnosed in the first two decades of life (mean age at diagnosis, 10 years). A few unequivocal cases of Coats disease have been newly diagnosed in older adults. It has no apparent preference for race but has a clear predilection for gender, with >75% of cases occurring in males. Most affected patients have unilateral disease, with approximately 95% of cases affecting only one eye and 5% affecting both eyes. When both eyes are affected, the disease is usually asymmetric with the fellow eye having only minimal involvement.5
Genetic associations
Coats disease is mostly sporadic nonhereditary condition that is not associated with identifiable systemic abnormalities.5 A genetic cause for Coats disease has been proposed based on several observation: 1) The association of retinal telangiectasia with muscular dystrophy and deafness in one family6; 2) Occurrence of exudative retinopathy in a few members of families with retinitis pigmentosa7; 3) The case of a mother purported to have unilateral retinal telangiectasia and a son with Norrie disease.8 The exact molecular mechanisms underlying Coats disease remain to be elucidated. Several candidate gene mutations are described, including the Norrie disease protein (NDP), CRB1 and PANK2.6-8
Table 1: List of Less Common Genetically Determined Isolated or Syndromic Ocular Conditions
Dyskeratosis congenita
Facio–scapulo–humeral dystrophy
Turner syndrome
Incontinentia pigmenti
Kabuki syndrome
Norrie disease
Retinitis pigmentosa with coats-like retinopathy
Senior–loken syndrome
Pathophysiology
The pathogenesis of Coats disease is vague and probably follows a general sequence of vents: The telangiectatic blood vessels (presumably congenital) have more permeable endothelial cells. Hence, they leak lipoproteins into the retina, causing retinal edema. As this material accumulates, it eventually breaks through the external limiting membrane of the retina, causing a nonrhegmatogenous exudative retinal detachment. As further lipoproteinaceous material accumulates, the retinal detachment becomes bullous and lies immediately posterior to the lens.5
Clinical features
Symptoms
Coats frequently present in childhood with painless unilateral decreased vision, leukocoria, or strabismus. Between 60% and 70% of cases present in the first decade of life. Adults may present with either symptom identical to those present in children or a milder form of the disease with predominantly macular vascular anomalies such as those seen in type 1 idiopathic macular telangiectasia.5
Signs
The ophthalmoscopic features of Coats disease include retinal telangiectasia, intraretinal exudation, and exudative retinal detachment. The retinal telangiectasia usually are located in the peripheral retina, most often between the equator and ora serrata inferotemporally.5,9 However, they can be confined to other quadrants or they may be widespread, affecting all quadrants. They are characterized by areas of generalized capillary dilation with small aneurysms that can cause focal enlargement of these dilated capillaries (Leber miliary aneurysms). These aneurysmal dilations, most commonly found surrounding areas of capillary dropout, tend to be radially oriented. In approximately one third of cases, these vascular changes extend posterior to the equator toward the retinal vascular arcades. Telangiectasia in the macular area is uncommon, occurring in <5% of cases.5 Occasionally, there are aneurysmal dilatations of the major retinal arteries and veins as well.
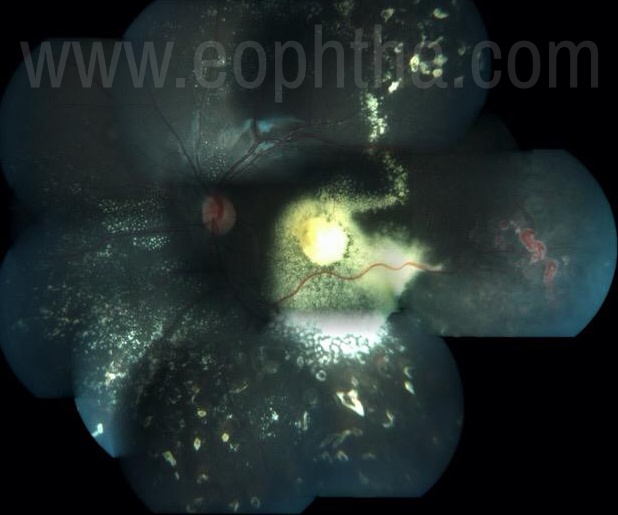
The exudation usually appears as areas of golden yellow lipoproteinaceous deposits in the sensory retina.5,9 It is generally more widespread than the telangiectasias and involves the retina diffusely in 75% of cases, often affecting areas remote from the main vascular abnormalities (with a particular predisposition for the macular region). There is no clear explanation for the exudation to preferentially accumulate in the macular area (Fig 1). When exudate accumulates in Henle’s layer (parafoveal outer plexiform layer), a macular star develops. Macular edema from posterior or peripheral vascular leakage is a common cause of visual loss in Coats disease. In the posterior pole, subretinal accumulations of exudate may develop, forming yellow-white mounds and broad sheets of lipid material primarily composed of cholesterol.10,11 Longstanding subretinal exudates may exhibit superficial crystalline deposits.10 Retinal blood vessels overlying patches of exudate may gradually become masked and undergo gliotic sheathing9 as the mass of exudate increases. Retinal pigment epithelial cells that proliferate and migrate into the subretinal space and undergo fibrous metaplasia have been associated with subretinal fibrous proliferation and resultant retinal detachment.
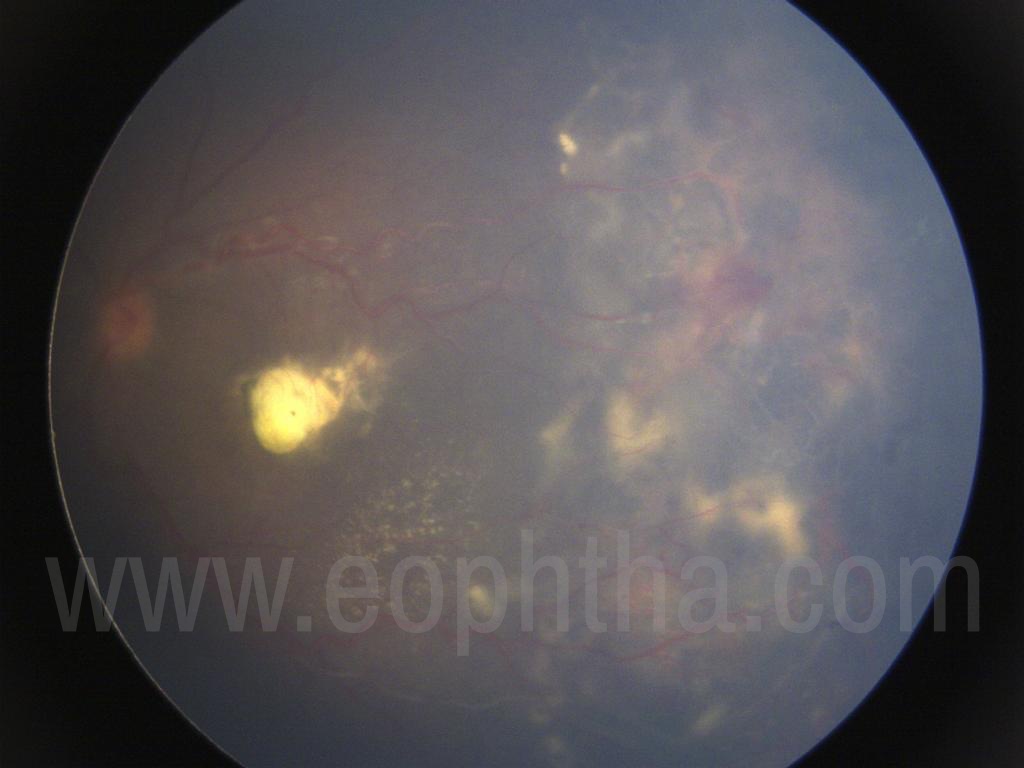
Patients with untreated Coats disease eventually develop a nonrhegmatogenous exudative retinal detachment. It generally begins in the areas of telangiectasia and slowly progresses to involve the entire retina. The extent of serous detachment increases as the vascular and exudative components of the disorder proceed, until a total retinal detachment overlying a yellow-green subretinal mass results. The detachment can eventually lie immediately behind the posterior lens capsule (Fig 2) and can progress to cause anterior displacement of the lens–iris diaphragm and secondary glaucoma.5 In few patients, the detachment spontaneously regresses before neovascular complications arise. Secondary complications include uveitis, glaucoma, and cataract.
The choroid and sclera appear to be unaffected by the primary pathology of Coats disease. The vitreous usually remains clear unless hemorrhage, neovascularization, massive leakage, or retinal breaks are present. Vitreoretinal traction, fibrosis, or proliferative vitreoretinopathy are rare; epimacular membrane may be more common than previously recognized, however.11 Preretinal fibrosis and vitreoretinal traction can be helpful differentiating features in disorders with a Coats-like clinical picture, such as vasoproliferative tumor and their presence warrants consideration of additional diagnoses.
Natural course & complications
The natural course and complications of Coats disease is important as some devastating complications warrants enucleation. In rare instances, the telangiectasias have been known to spontaneously regress. In most cases, however, the disease is slowly progressive with increasing exudative retinal detachment. Perhaps the most devastating complication is neovascular glaucoma, which occurs in approximately 10% of affected eyes.5 Patients with neovascular glaucoma often require primary enucleation, stressing the importance of early diagnosis and prompt treatment before the development of extensive retinal detachment.
In some instances, a child with Coats disease can develop signs of orbital cellulitis, presumably secondary to necrosis from elevated intraocular pressure and release of toxic substances in advanced disease. Another unusual complication of Coats disease is anterior chamber cholesterolosis, which occurs in approximately 3% of cases.5 The anterior chamber cholesterol apparently passes from the subretinal space through a peripheral retinal dialysis secondary to pressure from the subretinal fluid near the ora serrata.
Retinal bleeding from the abnormal blood vessels is somewhat unusual, occurring in approximately 13% of cases. Appreciable vitreous hemorrhage is extremely rare as a presenting feature of Coats disease. Neovascularization of the optic disk and retina occur in only 3% of cases. In view of the fact that Coats disease is characterized by retinal ischemia in the areas of telangiectasia, it is surprising that the incidence of neovascularization is so low. Intraretinal macrocysts occur in approximately 12% of eyes, particularly in areas of long-standing retinal detachment. They generally portend a poor visual prognosis. Secondary retinal vasoproliferative tumor develops in approximately 5% of patients with Coats disease.5 It is a vascular fundus mass that probably represents a proliferative response to chronic retinal detachment and retinal ischemia.
Classification
Different authors have proposed various classifications of Coats disease, although none is universally employed. Each system is based upon grading the clinical appearance of telangiectasia vessels or exudation leading to retinal detachment and devastating secondary ocular complications.
|
Table 2: Classifications For Coats Disease |
||
|
STAGE |
SHIELDS |
GOMEZ MORALES |
|
1 |
Telangiectasia only |
Isolated focal exudate |
|
2 |
Telangiectasia and Exudation
|
Massive elevated exudation |
|
3 |
Exudative retinal detachment
|
Partial retinal detachment |
|
4 |
Total retinal detachment and glaucoma |
Total retinal detachment |
|
5 |
Advanced end-stage disease |
Secondary complications (uveitis, glaucoma, cataract) |
Diagnosis:
Clinching features
Coats disease is clinical diagnosis, patient presenting in first decade of life with history of painless unilateral decreased vision, leukocoria, or strabismus and on ophthalmoscopic examination noted to have retinal telangiectasias, intraretinal exudation, and / or exudative retinal detachment.
Ancillary testing
Ancillary studies like ultrasonography, fluorescein angiography, Optical coherence tomography, radiological imaging and cytologic analysis of subretinal fluid can be helpful in substantiating the diagnosis in atypical cases where the diagnosis is more difficult.
Ophthalmic B-scan ultrasonography is useful, especially in clinical situations where posterior segment examination is limited by media opacity or poor patient cooperation. Ultrasonography can confirm the disease extent and most importantly rule out any potential malignancy by looking for any calcification or choroidal mass lesion.
Fluorescein angiography can delineate irregularly dilated, tortuous blood vessels and adjacent areas of retinal capillary dropout that characterize Coats disease. The vessels typically fill with fluorescein in the late arterial or early venous phase and show progressive leakage of dye into the adjacent retina and subretinal space. In late-phase angiograms, there is persistent confluent hyperfluorescence of the exudate in the retina and subretinal space. (Fig 3)
Optical coherence tomography (OCT) is useful in identifying subtle macular edema or cystic changes and to monitor response to treatment. OCT’s role in Coats disease parallels those for other retinal disorders in the era of anti-VEGF therapy. (Fig 4)
Radiologic imaging of the globe and orbit may be obtained with computed tomography (CT) and magnetic resonance imaging (MRI). CT scan is not preferred due to radiation exposure. MRIs demonstrate hyperintense T1- and T2- weighted signal converging on the optic nerve head corresponding to an exudative retinal detachment. This is in contrast to retinoblastoma, which is relatively hypointense on T2-weighted images. (Fig 5) After contrast administration, retinoblastoma reveals mass-like enhancement, in contrast to advanced Coats disease, which demonstrates linear enhancement convergent on the optic nerve head corresponding to detached retina.12
Cytologic analysis of subretinal fluid in Coats disease can confirm the diagnosis in cases where the diagnosis remains in question. This procedure should generally not be performed in cases where retinoblastoma is a reasonable diagnostic consideration. In Coats disease, cytopathologic studies disclose characteristic lipid-laden macrophages and cholesterol crystals.13
Differential diagnosis
It is extremely important for the clinician to recognize the characteristic clinical features of Coats disease and to differentiate it from other abnormalities that can be quite similar.
Table 3: Differential Diagnosis of Coats Disease
Tumors
Retinoblastoma
Hamartomas (Capillary or cavernous hemangioma)
Vasoproliferative tumors
Amelanotic melanoma
Inherited and congenital vitreoretinal disorders
FEVR
Norrie disease
Persistent fetal vasculature
Retinitis Pigmentosa
Idiopathic Retinal vascular disease and inflammation
Macular telangiectasia
Uveitis (Sarcoidosis; Tuberculosis)
Retinal vasculitis (Eales disease; IRVAN)
Sickle cell retinopathy
FEVR: Familial exudative vitreoretinopathy, IRVAN: Idiopathic retinal vascular aneurysm and neuroretinitis
Modalities of treatment:
Vascular Ablation:Thermal Laser Photocoagulation
Photocoagulation early in Coats disease, first described by Meyer-Schwickerath, has historically been the most efficacious treatment.14 The goal in Coats disease is to obliterate abnormal vasculature and hyper permeable aneurysmal dilations. Therefore, the absorptive property of hemoglobin to 532 nm (green) laser light is adequate for vascular absorption of laser energy, and this wavelength can be utilized for direct photocoagulation of vascular anomalies even in the presence of subretinal fluid. Infrared laser wavelengths have inadequate absorption spectra for intravascular chromophores. The effectiveness of the treatment may decrease when more than two quadrants of the retina exhibit vascular abnormalities. Close follow-up is required following ablative therapy, as late recurrences may require additional treatment.11
Cryotherapy
Telangiectatic vessels in the far peripheral retina in the setting of total exudative retinal detachment may preclude adequate laser photocoagulation; therefore, cryotherapy has been traditionally used in this setting. Triple freeze / thaw method is used i.e. Cryotherapy is used under indirect ophthalmoscopy and deployed until a white freeze reaction is observed thrice. There is some evidence that cryotherapy increases epiretinal membrane formation and retinal traction.15
AntiVEGF therapy:
A number of recent reports address the role of vascular endothelial growth factor (VEGF) in Coats disease and the use of intravitreal anti-VEGF compounds as an adjunct or single therapy. Intravitreal bevacizumab (Avastin, Genentech, South San Francisco, CA) is highly effective in decreasing edema and exudate and may even induce resolution of total retinal detachment after a single administration. Sun et al16 and Kaul et al17 demonstrated clinical improvement and decreased VEGF levels after pegaptanib therapy. Ramasubramanian et al18 reported 100% resolution subretinal fluid, 100% improvement in telangiectasias, 75% resolution of exudate, no retinal breaks but also found that there in increase in vitreoretinal fibrosis and TRD. The overwhelming weight of evidence, therefore, supports the use of anti-VEGF therapy for Coats disease with retinal edema or exudate involving the macula or when adequate ablative therapy cannot be used.
Surgical Management:
Surgery should be considered as a last resort for intractable total retinal detachments, a situation less often encountered in the era of anti-VEGF therapy. Surgical intervention may be required in cases with advanced Coats disease where photocoagulation and/or cryotherapy may not halt progression or significant epiretinal traction from preretinal membranes or proliferative vitreoretinopathy.19,20
Eyes with Coats disease may have an abnormal transretinal pressure gradient, because the retinal pigment epithelium capacity to maintain a fluid-free subretinal space may be overwhelmed by the exudative process. Therefore, internal drainage retinotomies should be avoided. Retinal breaks may lead to a persistent total exudative/rhegmatogenous retinal detachment as a result of the underlying RPE deficiency. Transscleral subretinal fluid drainage may then be required.
For severe Coats disease (stage 4 and / or 5 disease), subretinal drainage combined with vitreous infusion and ablation of abnormal vasculature can preserve the globe. As a result, patients with Coats can experience normal orbital growth and not suffer the cosmetic and psychological side effects associated with enucleation. Off lately with the advent of antiVEGF role of surgical management is very limited.
|
TABLE 4: Approach to management of Coats disease |
|||
|
STAGE |
TREATMENT |
FUNCTIONAL PROGNOSIS |
ANATOMICAL PROGNOSIS |
|
Telangiectasia only |
Observe / LPC |
Good |
Salvable |
|
Telangiectasia and Extrafoveal Exudation |
LPC / TCC |
Good |
Salvable |
|
Telangiectasia and Foveal Exudation |
TCC / LPC ±AntiVEGF |
Fair |
Salvable |
|
Subtotal Exudative retinal detachment |
TCC / LPC ±AntiVEGF |
Poor |
Salvable |
|
Total Exudative retinal detachment |
Surgery ±AntiVEGF |
Poor |
± |
|
Total retinal detachment and glaucoma |
Enucleation |
Nil |
Non-Salvable |
|
Advanced end-stage disease |
Observe |
Nil |
Non-Salvable |
VEGF: Vascular endothelial growth factor
References
1. Coats G. Forms of retinal diseases with massive exudation. Roy Lond Ophthalmol Hosp Rep 1908;17:440–525.
2. von Hippel E. Ueber eine seltene erkrankung der netzhaut. Arch f Augenh. 1904;59:83
3. Leber T. Ueber eine durch Vorkommen multipler Miliaraneurysmen charakterisierteForm von Retinaldegeneration.
Albrecht von Graefe’s Arch Klin Ophthalmol 1912;81:1–14.
4. Reese AB. Telangiectasis of the retina and Coats disease. Am J Ophthalmol 1956;42:1–8.
5. Shields JA, Shields CL, Honavar S, Demirci H. Coats disease. Clinical variations and complications of Coats disease
in 150 cases. The 2000 Sanford Gifford Memorial Lecture. Am J Ophthalmol 2001;31:561–571.
6. Small RG. Coats disease and muscular dystrophy. Trans Am Acad Ophthalmol Otolaryngol 1968;71:225–231.
7. Khan JA, Ide CH, Strickland MP. Coats’-type retinitis pigmentosa. Surv Ophthalmol. 1988;32:317e32
8. Black GC, Perveen R, Bonshek R, et al. Coats’ disease of the retina (unilateral retinal telangiectasis) caused by somatic mutation in the NDP gene: a role for Norrin in retinal angiogenesis. Hum Mol Genet. 1999;8:2031e5
9. Theodossiadis GP. Some clinical, fluorescein-angiographic, and therapeutic aspects of Coats disease. J Pediatr Ophthalmol Strabismus 1974;16:257–262.
10. Duke JR. The role of cholesterol in the pathogenesis of Coats disease. Trans Am Ophthalmol Soc. 1963;61:492e544
11. Shields JA, Shields CL, Honavar SG, et al. Classification and management of Coats disease: the 2000 Proctor Lecture. Am J Ophthalmol. 2001;131:572e83
12. Eisenberg L, Castillo M, Kwock L, et al. Proton MR spectroscopy in Coats disease. AJNR Am J Neuroradiol. 1997;18:727e9
13. Shields JA, Shields CL, Ehya H, Eagle RC Jr, DePotter P. Fine needle aspiration biopsy of suspected intraocular tumors. The 1992 Urwick Lecture. Ophthalmology 1993;100:1677–1684.
14. Pesch KJ, Meyer-Schwickerath G. [Light coagulation in morbus Coats and Leber’s retinitis]. Klin Monbl Augenheilkd. 1967;151:846e53
15. Glaser BM, Vidaurri-Leal J, Michels RG, et al. Cryotherapy during surgery for giant retinal tears and intravitreal dispersion of viable retinal pigment epithelial cells. Ophthalmology. 1993;100:466e70
16. Sun Y, Jain A, Moshfeghi DM. Elevated vascular endothelial growth factor levels in coats disease: Rapid response to pegaptanib sodium. Graefes Arch Clin Exp Ophthalmol. 2007;245:1387e8
17. Kaul S, Uparkar M, Mody K, et al. Intravitreal anti-vascular endothelial growth factor agents as an adjunct in the management of coats’ disease in children. Indian J Ophthalmol. 2010;58:76e8
18. Ramasubramanian A, Shields CL. Bevacizumab for Coats’ disease with exudative retinal detachment and risk of vitreoretinal traction. Br J Ophthalmol. 2012;96:356e9
19. Mrejen S, Metge F, Denion E, et al. Management of retinal detachment in Coats disease. Study of 15 cases. Retina. 2008;28:S26e32
20. Shukla D, Chakraborty S, Behera UC, et al. Vitrectomy for epimacular membrane secondary to adult-onset coats’ disease. Ophthalmic Surg Lasers Imaging. 2008;39:239e41