.jpeg)

Brief Scenario of the case:
A 25 year-old-female, resident of Rajasthan presented to us with the chief complaints of diminution of vision in the right eye for a period of 3 days and redness with mild pain in both eyes for one month.
History of Present Illness:
The patient was apparently well 3 days back when she started to develop diminution of vision in her right eye. She had mild pain and redness in both her eyes for the last one month.
Past Ocular History:
She was diagnosed as a case of right eye choroiditis elsewhere. Routine blood counts were normal. Erythrocyte sedimentation rate (ESR) was 22mm in the first hour. Veneral Disease Research Laboratory (VDRL) test was negative. Human Immunodeficiency Virus (HIV) test was negative.
Past Medical History/ History of Medication:
The patient was using topical steroids and cycloplegic eye drops. No history of tuberculosis or any systemic disease.
Family History:
There was no significant family history.
Review of Systems/Systemic Examination:
The systemic examination appeared to be within normal limits.
OCULAR EXAMINATION:
Best Corrected Visual Acuity (Snellen)
- Right eye (OD): Counting fingers at 2 meters
- Left eye (OS): 6/9
Ocular Motility/Alignment: Full free and painless
Intraocular Pressure (IOP)
- OD: 10 mmHg
- OS: 10 mmHg
Pupils: dilated due to use of cycloplegics
Slit-lamp examination:
- Lids/lashes: normal both eyes
- Conjunctiva/sclera: circumciliary congestion in both eyes
- Cornea: clear in both eyes
- Anterior chamber: deep and flare 3+ , cells 3+ in both eyes
- Iris: within normal limits in both eyes
- Lens: iris pigments present on anterior capsule of lens in right eye and clear in left eye.
Dilated Fundus examination (Right eye)
- Vitreous: Vitritis 2+ was present
- Disc: edema was present
- Macula: foveal reflex was absent
- Vessels: dilated tortuous
- Multiple subretinal fluid (SRF) pockets and exudative detachment was present.
Dilated Fundus examination (Left eye)
- Vitreous: Vitritis 1+ was present
- Disc: edema was present
- Macula: foveal reflex was absent, choroidal folds present
- Vessels: dilated tortuous
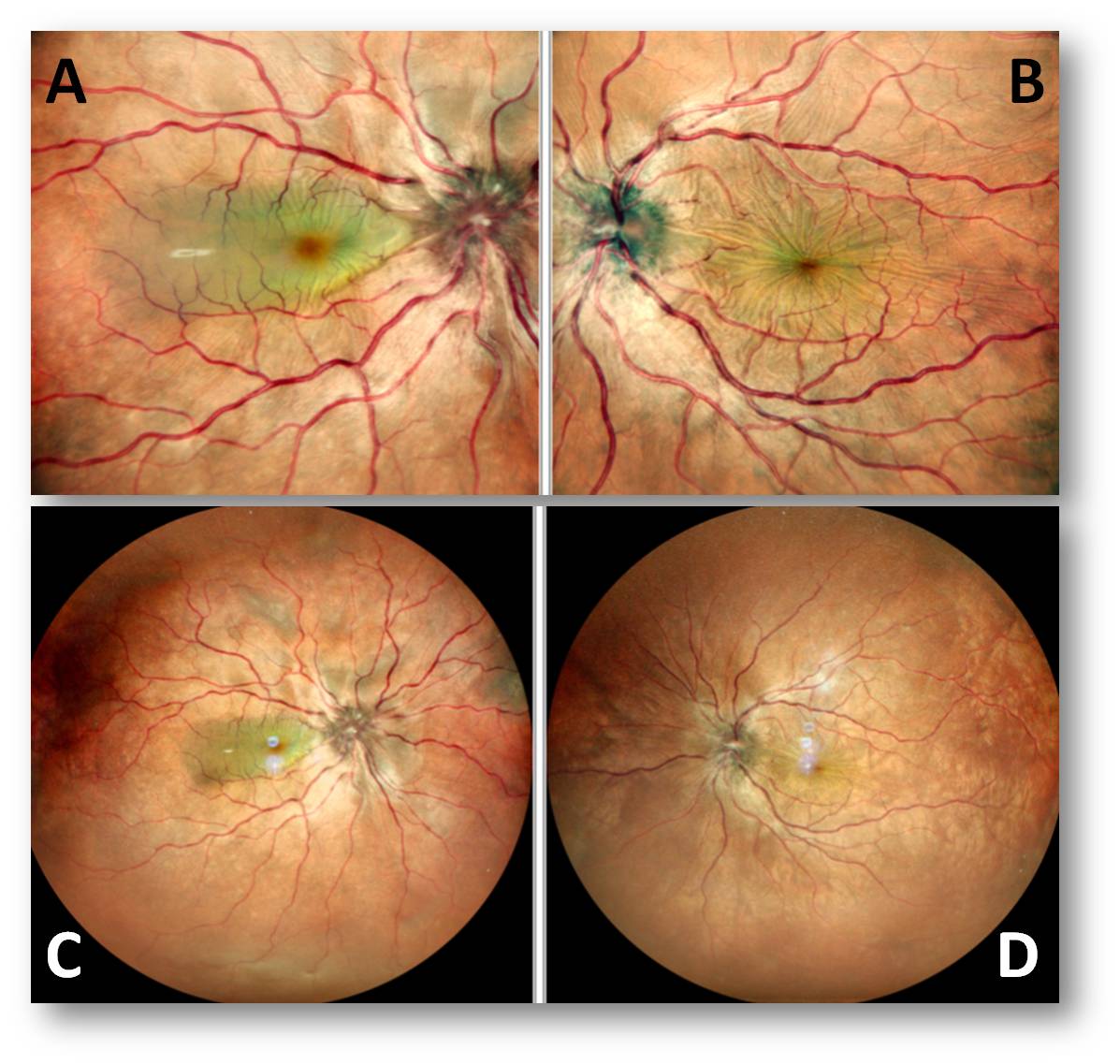
Ancillary Investigations:
Fundus photograph of the right eye (Figure 1 A and C) showed disc edema with multiple subretinal pockets. Left eye (Figure 1 B and D) showed disc edema with choroidal folds.
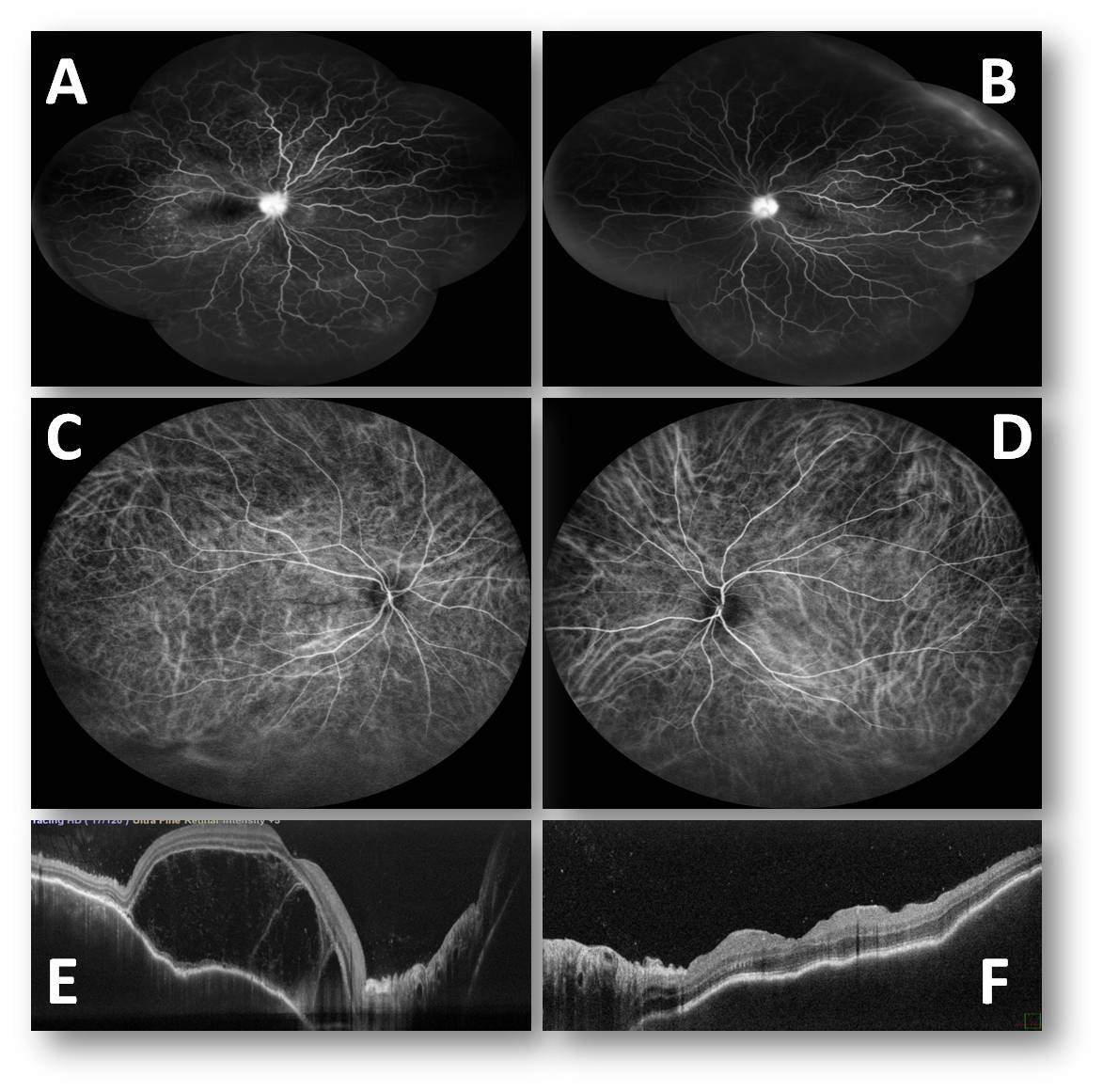
Fundus Fluorescein Angiography (FFA) (Figure 2A and 1B) of both the eyes showed multiple pinpoint areas of hyperfluorescence,, diffuse vascular leakage with disc leakage.
Indocyanine green angiography (Figure 2C and 2D) showed multiple hyperfluorescent dark spots in the mid-phase of the angiography.
OCT (Optical coherence Tomography) of the right eye (Figure 2E) showed multiple subretinal fluid pockets, retinal pigment epithelial irregularity, choroidal thickening, and vitreous cells. OCT of the left eye (Figure 2F) showed retinal pigment epithelial layer irregularity with choroidal thickening.
Differential Diagnosis:
- Vogt Koyanagi Harada (VKH) Disease
- Intraocular Lymphoma
- Central Serous Retinopathy
- Sympathetic Ophthalmia
- Sarcoidosis
Diagnosis:
OU Vogt Koyanagi Harada Disease
Treatment:
Intravenous injections of Methylprednisolone were given for 3 days which was followed by oral steroids 1 mg per day for 10 days with taper 5 mg after 1o days. Topical steroids eye drop with atropine was started in both eyes.
She was also started on tab azathioprine 50mg twice a day after systemic evaluation in consultation with a rheumatologist. The patient was followed monthly.
Course of the disease:
After two months (Figure 3), her visual acuity improved to 6/9 and 6/6 in the right and left eye respectively. She was continued on a tapering course of steroids and azathioprine with routine systemic evaluation.
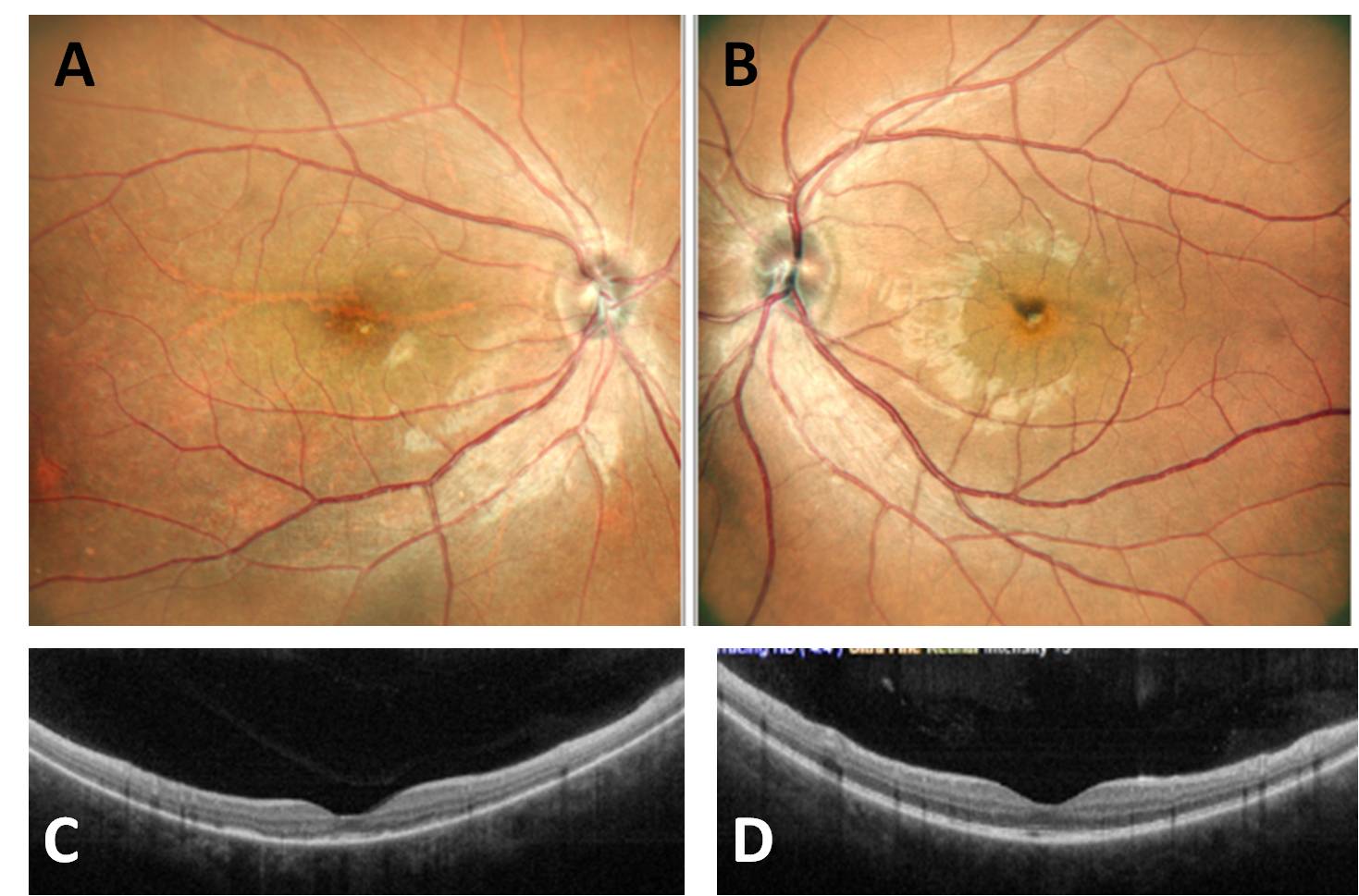
The patient did not present with any systemic signs like headache and tinnitus. During the course of the disease too she did not develop any systemic signs like alopecia, poliosis, or vitiligo. According to the diagnostic criteria for Vogt – Koyanagi – Harada’s disease she was a case of probable VKH disease.
Discussion:
Introduction
Vogt-Koyanagi-Harada (VKH) disease is an idiopathic multisystem autoimmune disease against melanocytes containing tissues such as uvea, ear, and meninges, and it is mainly mediated by cellular immune responses. [1]
Vogt-Koyanagi disease is characterized mainly by skin changes and anterior uveitis. In Harada disease, neurological features and exudative retinal detachments predominate. The choroid is the primary site of inflammation in Vogt- Koyanagi-Harada (VKH) disease. [2]
The clinical manifestations of VKH disease are panuveitis accompanied by neurological signs such as pleocytosis of the cerebrospinal fluid, skin signs such as alopecia and vitiligo, and internal ear disturbances.
The most commonly observed ocular changes detectable at the early stage of VKH disease are iridocyclitis, exudative retinal detachment, and swelling of the optic disc.
Epidemiology And Pathogenesis
Vogt-Koyanagi-Harada disease occurs more frequently in pigmented races, such as Asians, Hispanics, and Native Americans.
The disease affects women more frequently than men. Of the 65 patients with VKH who participated in one study, 78% were Hispanic, 10% were Asian, 6% were Black, and 3% were white; one patient was an Asian Indian and one was a Native American. [3] In this study, 74% were women. Most were in the second to fifth decades of life at the onset of illness. The age range was 7-71 years, with a mean of 32 years.
There is evidence of increased risk among those with certain HLA genotypes (DRB1 ∗0405). It indicates that there is a genetically determined susceptibility for VKH disease. [4][5] The underlying cause of the disease remains unknown.
Clinical Features
Clinically, the disease is categorized into four phases: prodromal, uveitic, chronic, and recurrent. [6]
Prodromal Phase
Patients may present with influenza-like symptoms with fever, headache, and sometimes nausea. Within 1-2 days of the onset of the prodromal phase, the patient begins to complain of blurred vision, photophobia, redness of the conjunctiva, and ocular pain. There are also complaints of sensitivity of the scalp, hair, and skin to touch during the prodromal phase.
Uveitic Phase
In the early uveitic phase, cells are present in the anterior chamber, the vitreous, or both. There is. thickening of the posterior choroid. There are multiple serous retinal detachments or small folds that radiate from the macula. In severe cases, the detachment may become bullous. The optic disc becomes hyperemic and edematous.
Chronic Phase
After treatment with systemic corticosteroids, the elevation of the neural retina gradually disappears as the subretinal fluid is absorbed. Cells in the anterior chamber and vitreous decrease or disappear.
After several months depigmentation may occur in the fundus, giving it a sunset-glow appearance. Small, discrete, and scattered depigmented lesions may be seen within the sunset-glow fundus. Most of these lesions represent degenerated or lost retinal pigment epithelium (RPE). [7]
Depigmentation in the corneal limbus is also sometimes noted about 1 month after the onset. This limbal depigmentation is known as Sugiura’s sign.
Recurrent Phase
In recurrent cases, anterior uveitis is more predominant than posterior uveitis. The eyes show signs of chronic iridocyclitis. Mutton-fat keratic precipitates, Koeppe iris nodules, and posterior synechiae are signs of the chronic recurrent phase. The iris becomes atrophic and loses some if its pigmentation.
Choroidal neovascularization (CNV) may occur in the peripapillary region and the macula often causing retinal hemorrhage. Retinal pigment epithelial proliferation may result in subretinal fibrosis.
SYSTEMIC ASSOCIATIONS
- Auditory Signs: They consist of sensorineural hearing loss with tinnitus and vertigo (usually present at the onset of the disease).
- Neurological Signs: May include fever, headache, neck stiffness, nausea and vomiting.
- Dermal Signs: Vitiligo can occur on the face, hands, shoulders and lower back about 2-3 months after the onset of VKH.
- Other Signs: Poliosis and alopecia are often present.
The revised diagnostic criteria for VKH disease were discussed at the First International Workshop on Vogt-Koyanagi-Harada disease and reported from the consensus committee.
Table 1. Diagnostic criteria for Vogt-Koyanagi-Harada’s disease
1. No history of penetrating ocular trauma or surgery preceding the initial onset of uveitis.
2. No clinical or laboratory evidence suggestive of other disease entities.
3. Bilateral ocular involvement in which either part a or b must be met, depending on the stage of the disease when the patient was examined:
a. Early manifestations of the disease –
(1) There must be evidence of a diffuse choroiditis (with or without anterior uveitis, vitreous inflammatory reaction or optic disk hyperemia) which may manifest as one of the following:
(a) Focal areas of subretinal fluid
(b) Bullous serous retinal detachment
(2) With equivocal fundus findings, both of the following must be present as well:
(a) Focal areas of delay in choroidal perfusion, multifocal areas of pinpoint leakage, large placoid areas of hyperfluorescence, pooling within the subretinal fluid, and optic nerve staining (listed in order of sequential appearance) by fluorescein angiography
(b)Diffuse choroidal thickening, without evidence of posterior scleritis by ultrasonography.
b. Late manifestations of the disease-
(1) History suggestive of prior presence of findings from 3a and either both 2 and 3 listed below or multiple signs from 3
(2) Ocular depigmentation (either of the following manifestations is sufficient)
(a) Sunset glow fundus
(b) Sugiura’s sign
(3) Other ocular signs (any of the following manifestations are sufficient)
(a) Nummular chorioretinal depigmentation scars
(b) Retinal pigment epithelium clumping and/or migration
(c) Recurrent or chronic anterior uveitis.
4. Neurological/auditory findings (which may have resolved by the time of examination). (Any of the following manifestations are sufficient)
a. Meningismus (malaise, fever, headache, nausea, abdominal pain, stiffness of the neck or back, or a combination of these factors. However, headache alone is not enough to meet a definition of meningismus)
b. Tinnitus
c. Cerebrospinal fluid (CSF) pleocytosis.
5. Integumentary findings (which do not precede the onset of the central nervous system or ocular disease). (Any of the following manifestations are sufficient)
a. Alopecia
b. Poliosis
c. Vitiligo.
Complete Vogt-Koyanagi-Harada’s disease–Criteria disease criteria 1–5 must be present
Incomplete Vogt-Koyanagi-Harada’s disease–Criteria disease criteria 1–3 and either 4 or 5 must be present
Probable Vogt-Koyanagi-Harada’s disease–Criteria disease isolated ocular disease-criteria 1–3 must be present.
ANCILLARY INVESTIGATION
FUNDUS FLUORESCEIN ANGIOGRAPHY:
In the early phase of VKH, fluorescein fundus angiography reveals numerous hyperfluorescent dots at the level of the RPE. These dots have a tendency to gradually enlarge. The dye leaks through the RPE and accumulates in the subretinal space.
In the chronic phase, angiography reveals diffusely scattered dots of hyperfluorescence due to the window defects at the level of the RPE.
Indocyanine green angiography:
ICG demonstrates an early choroidal stromal vessel hyperfluorescence and hypofluorescent dark spots during the early and mid-phase, distributed mainly posteriorly. During the active stage, the hypofluorescent spots fade and are replaced by hyperfluorescent ones (focal areas of active choroidal inflammation).
In the chronic stage, the hypofluorescent dark spots are seen during all the phases of ICG.
Optical Coherence Tomography:
It shows the presence of subretinal fluid. Multiple septae creating pockets of fluid in outer retina are seen. In the presence of choroidal folds, OCT shows corrugation of the RPE/choroid with choroidal thickening. Increased choroidal thickening can be appreciated in EDI (Enhanced Depth Imaging) OCT.
Ultrasonography
In the acute stage, the ultrasound shows diffuse choroidal thickening, serous retinal detachments, vitreous opacities and scleral or episcleral thickening.
Electroretinography
Full-field electroretinography (ERG) shows diffusely diminished amplitudes in both scotopic and photopic phases in the chronic stage.
Laboratory Investigation:
Cerebrospinal fluid examination shows pleocytosis (which may persist for up to 8 weeks)and elevation of protein levels in the early stages.
Treatment
Vogt-Koyanagi-Harada disease is treated with high-dose systemic steroids with a slow taper. Immunosuppressive therapy is added to prevent recurrences and in patients who could not tolerate steroids
In the early phase of the disease, high-dose corticosteroids are given intravenously or orally. Steroids are gradually tapered over a period of six to eight months. Lai TY et al9 reported that patients receiving treatment for less than 6 months were more likely to have recurrences (58.8%) compared to those treated for 6 months or more (11.1%). The immunosuppressants that have proven efficient are Azathioprine, Methotrexate, Cyclosporine, and Mycophenolate mofetil. It is important to remember that the administration of immunosuppressants should be closely monitored and evaluated alongside an internist for any complications or side effects from the treatment.
Topical corticosteroids and cycloplegics are administered until the cells in the anterior chamber disappear.
In case of recurrence combination of immunosuppressive drugs is given or biologicals are added.
Course And Outcome
Uveitis due to VKH disease needs a longer duration of treatment and regular follow-up. However, a posterior subcapsular cataractmay develop as a complication of prolonged uveitis and a side effect of corticosteroids. Other complications include secondary angle-closure glaucoma, subretinal neovascularization, and subretinal fibrosis.
Differential diagnosis
- Sympathetic ophthalmia – sympathetic ophthalmitis can present as panuveitis associated with exudative retinal detachment. But there is always a history of ocular trauma in the exciting eye. There is no gender predisposition and occurs at any age.
- Central serous chorioretinopathy (CSCR)- It is usually unilateral, more common in males with minimal visual loss. Multifocal CSCR may present with exudative retinal detachment. There are no vitreous or anterior chamber cells in CSCR. Fluorescein angiography does not show any disc hyperfluorescence and there are inkblot or smoke stack leakages. In OCT, folds of RPE, fluctuations of ILM, subretinal septa are seen in VKH and pigment epithelial detachments (PEDs) are seen more in CSCR. Stress is the major risk factor for CSCR. Use of steroids can induce CSCR. Most cases of central serous chorioretinopathy clear up in one or two months without any treatment.
- Uveal effusion syndrome- Itis common in nanophthalmic eyes and hypermetropic eyes with the thickened sclera. It is not normally associated with intraocular inflammation and responds poorly to systemic steroid
- Posterior scleritis- It is usually unilateral and associated with Tsign in ultrasonography (a hyporeflective layer of fluid in the Tenon space surrounding the nerve and high reflectivity of the thickened sclera). It is not associated with neurologic and dermatologic findings.
Vogt – Koyanagi - Harada disease |
|
Etiology :
|
Signs:
|
Symptoms:
|
Differentials :
|
Investigations :
|
Treatment :
|
References
- Kitamura M, Takami K, Kitachi N, et al. Comparative study of two sets of criteria for the diagnosis of Vogt-Koyanagi-Harada’s disease. Amer J Ophthal 2005;139(6):1080-6.
- Gieser AS, Murphy RP. Eales’ disease. In: Ryan SJ (Ed). Retina, 2nd edition. St. Louis: CV Mosby; 1994.pp.1503-7.
- Moorthy R.S.,Inomata H.,Rao N.A.:Vogt-Koyanagi-Harada syndrome. Surv Ophthalmol1995;39:265-292.
- Shindo Y.,Inoko H.,Yamamoto T.,Ohno S.:HLA-DRB1 typing of Vogt-Koyanagi-Harada’s disease by PCR-RFLP and the strong association with DRB1∗0405 and DRB1 ∗0410. Br J Ophthalmol1994;78:223-226.
- Shindo Y.,Ohno S.,Nakamura S.,et al:A significant association of HLA-DRB1∗0501 with Vogt-Koyanagi-Harada’s disease results from a linkage disequilibrium with the primarily associated allele, DRB1∗0405. Tissue Antigens1996;47:344-345.
- Sigiura S.:Vogt-Koyanagi-Harada disease. Jpn J Ophthalmol1978;22:229-242.
- Inomata H.,Rao N.A.:Depigmented atrophic lesions in sunset glow fundi of Vogt-Koyanagi-Harada disease. Am J Ophthalmol2001;131:607-614.
- Lai, T., Chan, R., Chan, C.et al.Effects of the duration of initial oral corticosteroid treatment on the recurrence of inflammation in Vogt-Koyanagi-Harada disease.Eye23,543–548 (2009).