
Corneal dystrophies are classically defined as a group of inherited disorders of the cornea which is bilateral, symmetric, slowly progressive, and not related to any environmental or systemic factors.1,2 Most of the corneal dystrophies begin in the early decades of life and progress gradually. The effect of these changes in some of the dystrophies may not be clinically apparent till the later years. The underlying basis of all corneal dystrophies are genetic mutations some of which have been mapped, while others are yet to be identified.1-3 Most of these mutations result in the transcription of aberrant proteins. These proteins are deposited in various corneal layers and give rise to the characteristic clinical features. The pace of deposition also determines the natural history of the corneal dystrophy.
Corneal dystrophies were first reported by Groenouw in his article published in 1890.1 He described one patient with granular corneal dystrophy and another patient with macular corneal dystrophy using the term “cornea noduli” for both the conditions. Since then many reports of single or groups of cases have been published in ophthalmic literature describing different types of corneal dystrophies. Over the years, these descriptions varied or were supplemented by newer features obtained from advances in ophthalmic imaging technique; evolving from the pre- slit-lamp to the slit-lamp era and now confocal microscopy, light microscopic to electron-microscopy examinations, and finally, to genetic typing. The overlapping or incomplete manifestation of corneal signs due to incomplete gene penetrance, early presentation in the natural course of the dystrophies when all the signs have not developed and the use of eponyms are some of the reasons that have led to mis-classification of dystrophies or the same dystrophy getting named differently. The advancements in genetics and molecular biology have led to a better understanding of the underlying pathological mechanisms. It is evident now that multiple genes could give rise to a single corneal dystrophy with similar clinical features (genetic heterogeneity), or a single gene could result in multiple corneal dystrophies with separate and distinct clinical features (phenotypic heterogeneity).3
In 2005 the International Committee for Classification of Corneal Dystrophies (IC3D) was created to revise the nomenclature of corneal dystrophies.1 The intention was to accurately report the clinical and histological features, and genetic characteristics based on current evidences,1 which have been periodically upadated.2 The IC3D developed templates that included updated genetic, pathological and clinical information with representative clinical photographs. It also categorized the level of evidences supporting the existence of a dystrophy into 4 categories, with Category 1 having the highest evidence for denoting a phenotypic presentation as a corneal dystrophy and Category 4 having the least convincing evidence.1,2
Traditionally the corneal dystrophies have been classified according to the anatomical location of the lesions – epithelial, Bowman’s layer, stroma and endothelium.4,5 However, certain dystrophies extend beyond these strict anatomical demarcations like the Reis-Buckler Corneal Dystrophy, which initially affects the sub-epithelial area and Bowman’s layer and later the anterior and deep stroma.2 The current IC3D classification replaces the traditional classification system with a new system: epithelial and subepithelial dystrophies, epithelial-stromal TGFBI dystrophies, stromal dystrophies, and endothelial dystrophies (Table 1).2
|
Layer |
Name |
Abbreviation |
Category |
|
Epithelial & subepithelial dystrophies |
Epithelial basement membrane dystrophy |
EBMD |
1 |
|
Epithelial recurrent erosion dystrophies |
ERED |
3 |
|
|
Subepithelial mucinous corneal dystrophy |
SMCD |
4 |
|
|
Meesmann corneal dystrophy |
MECD |
1 |
|
|
Lisch Epithelial corneal dystrophy |
LECD |
2 |
|
|
Gelatinous droplet-like corneal dystrophy |
GDLD |
1 |
|
|
Epithelial-stromal TGFB1 dystrophies |
Reis-Buckler’s corneal dystrophy |
RBCD |
1 |
|
Thiel-Bienke corneal dystrophy |
TBCD |
1 |
|
|
Lattice corneal dystrophy type 1 |
LCD1 |
1 |
|
|
Granular corneal dystrophy type 1 |
GCD1 |
1 |
|
|
Granular corneal dystrophy type 2 |
GCD2 |
1 |
|
|
Stromal dystrophies |
Macular corneal dystrophy |
MCD1 |
1 |
|
Schnyder corneal dystrophy |
SCD |
1 |
|
|
Congenital stromal corneal dystrophy |
CSCD |
1 |
|
|
Fleck corneal dystrophy |
FCD |
1 |
|
|
Posterior amorphous corneal dystrophy |
PACD |
1 |
|
|
Central Cloudy dystrophy of Francois |
CCDF |
1 |
|
|
Pre-Descemet corneal dystrophy |
PDCD |
1 or 4 |
|
|
Endothelial dystrophies |
Fuchs endothelial corneal dystrophy |
FECD |
1, 2, or 3 |
|
Posterior polymorphous corneal dystrophy |
PPCD |
1 or 2 |
|
|
Congenital hereditary endothelial syndrome |
CHED |
1 |
|
|
X-linked endothelial corneal dystrophy |
XECD |
2 |
Table 1. The IC3D Classification2
Category 1: A well-defined corneal dystrophy in which the gene has been mapped and identified and the specific mutations are known.
Category 2: A well-defined corneal dystrophy that has been mapped to one or more specific chromosomal loci, but the gene(s) remains to be identified.
Category 3: A well-defined corneal dystrophy in which the disorder has not yet been mapped to a chromosomal locus.
Category 4: This category is reserved for a suspected, new, or previously documented corneal dystrophy, although the evidence for it, being a distinct entity is not yet convincing.
Description of corneal dystrophies
The following section gives a brief description of some common corneal dystrophies. The details are categorized according to the current IC3D templates. 2 An interested reader may refer the original publication for a more comprehensive review.
Epithelial and subepithelial dystrophies
|
|
Epithelial basement membrane dystrophy |
Meesmann corneal dystrophy |
Gelatinous droplet-like corneal dystrophy |
|
Eponyms |
Map-dot-fingerprint dystrophy Cogan microcystic epithelial dystrophy Anterior basement membrane dystrophy |
Juvenile hereditary epithelial dystrophy; Stocker-Holt variant |
Subepithelial amyloidosis or Primary familial amyloidosis (Grayson) |
|
Inheritance |
Uncertain |
Autosomal dominant |
Autosomal recessive |
|
Genetic locus |
5q31 |
Locus 12q13 (KRT3) Locus 17q12 (KRT12) Stocker–Holt variant |
1p32 |
|
Gene |
Transforming growth factor beta-induced (TGFBI) in 2 families |
Keratin K3 Keratin K12 – Stocker-Holt variant |
Tumor-associated calcium signal transducer 2 (TACSTD2, previously M1S1). |
|
Onset & course |
Adult; rare in children. Fluctuate with time. |
Early childhood. Stationary or slowly progressive |
1st to 2nd decade. Progressive. There is a possibility of recurrence of dystrophy after keratoplasty. |
|
Symptoms |
Asymptomatic or recurrent corneal erosions – pain, lacrimation & blurred vision. |
Asymptomatic or mild blurring of vision with glare and photosensitivity. Foreign body sensation to frank erosions may occur |
Significant photophobia, foreign body sensation, redness, tearing and decrease in vision |
|
Signs |
Irregular geographic grayish white superficial corneal lesions with distinct scalloped borders(maps, Figure 1) alone or in combination with irregular round, or oval non-staining whitish gray opacities single or clustered (dots); or parallel curvilinear lines in the paracentral cornea best visualized against the red glow of the slit-lamp retro-illumination(finger-prints); or ground-glass appearance under retro-illumination(bleb pattern). |
Multiple tiny intra-epithelial vesicles extend to the limbus and are more numerous in the interpalpebral area with clear surrounding epithelium. Whorled, wedged shaped opacities, cyst-like lesions or refractile linear opacities. In the Stocker-Holt variant the entire corneal is covered by fine grayish opacities that are fluorescein positive with fine linear lines in whorl pattern |
Initially sub-epithelial lesions appear similar to band keratopathy that progresses to small, multiple grayish nodules(mulberry lesions, Figure 2). The epithelium may stain with fluorescein. Superficial vascularization is present. In advanced cases there is stromal opacities with large nodules and the cornea resembles the kumquat fruit (kumquat-lesions). |
|
Pathology |
Sheets of intra-epithelial, multi-lamellar, basal laminar material (maps); intra-epithelial extensions of basal laminar material(finger-print lines); intra-epithelial pseudocysts containing cytoplasmic material (dots) & subepithelial accumulation of fibrillar-granular material (blebs) |
Presence of intra-epithelial cyst with PAS-positive cellular debris. |
Deposition of sub-epithelial and stromal amyloid. |
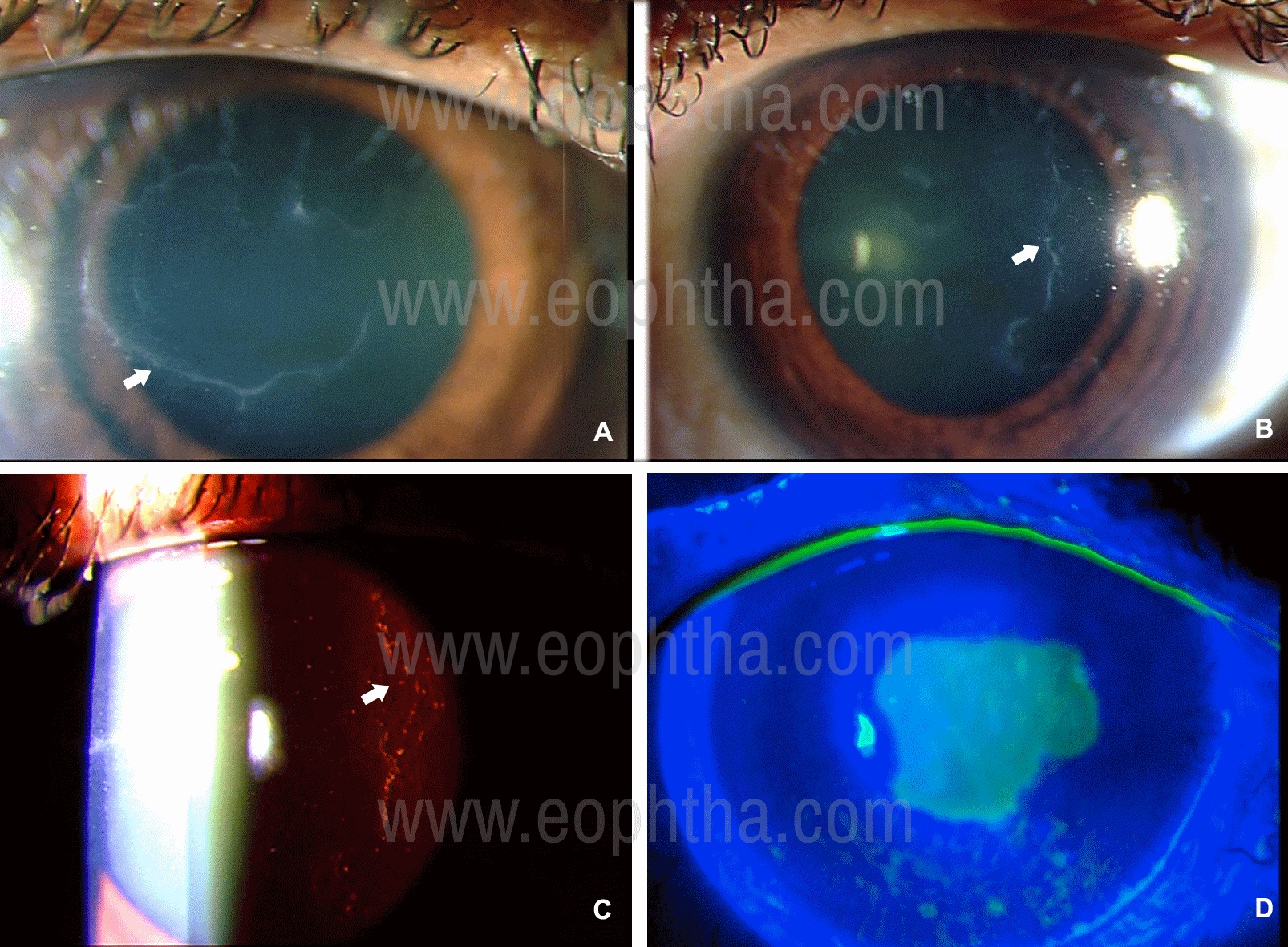
Figure 1: Slit-lamp photographs of Epithelial Basement Membrane Corneal Dystrophy (A-D) in a 30-year old lady. Map like changes are visible (A) and in fundus retro-illumination intra-epithelial dot-like opacities are seen which are either discreet or crowded as blebs (C). Fluorescein staining of the epithelium under blue-light illumination reveals a large epithelial defect as a result of corneal erosion (D).
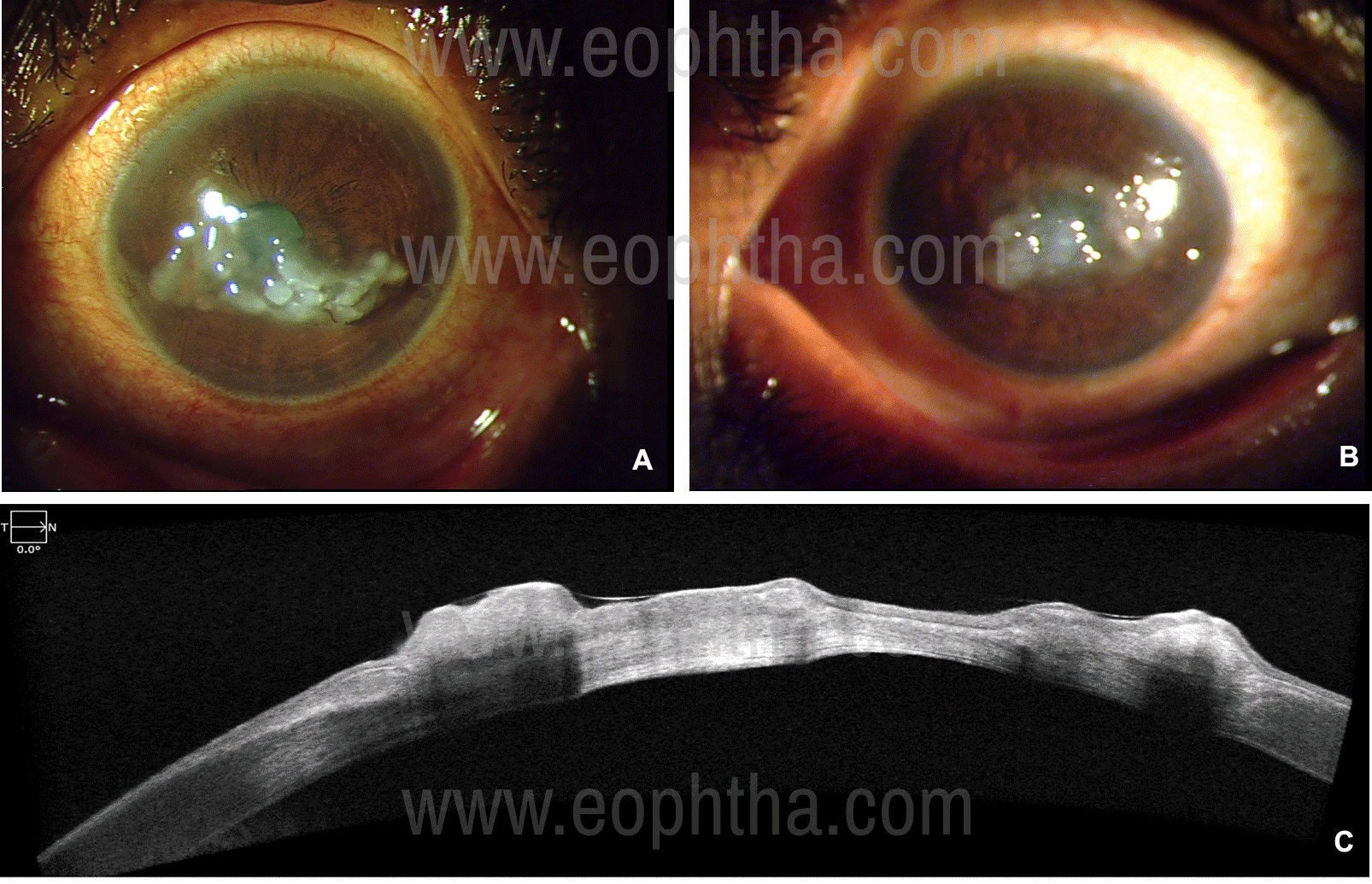
Figure 2: Slit-lamp photographs of Gelatinous drop-like corneal dystrophy in a 59-year old lady who presented with ocular irritation and photophobia. The photographs show the mulberry type of lesions present in both the eyes (A&B). Anterior segment optical coherence tomography picture of the right eye (C) shows dense, hyper-reflective, nodular formation in the epithelium with underlying optical shadowing. The stroma is hyper-reflective in the center indicating diffuse scarring till the endothelium.
Treatment:
Epithelial basement membrane dystrophy & Meesmann corneal dystrophy6
- Asymptomatic patients do not require any treatment.
- For foreign body sensation, ocular irritation frequent preservative-free lubricating eye drops.
- For acute corneal erosions: Broad-spectrum topical antibiotics and frequent preservative-free lubricating eye drops. If the patient is highly symptomatic then overnight patching with broad-spectrum antibiotic ointment and cycloplegic can be done. 6,7 Some advocate the use of the hypertonic saline solution or eye drops (5% sodium chloride) during day and ointment at night. High oxygen transmissibility bandage contact lens can also be used. One must be very judicious in prescribing bandage contact lenses because of the risk of secondary microbial keratitis. Patient hygiene and close follow-up/reviews should be ensured, and prophylactic broad-spectrum antibiotics should be concomitantly prescribed in patients treated with bandage contact lenses.
- For recurrent corneal erosions: Epithelial debridement of the involved area under strict asepsis with topical broad-spectrum antibiotics and preservative-free lubricating eye drops. The surface can also be polished with a diamond burr. Excimer laser phototherapeutic keratectomy is an alternative. These procedures may need to be repeated.
- A combination of chloramphenicol and polymyxin B is a good example of a broad-spectrum antibiotic.
Gelatinous droplet-like corneal dystrophy 6,8
- In early stages conservative management with preservative-free lubricating eye drops.
- If the nodules are large then superficial keratectomy with or without amniotic membrane grafting can be attempted. An alternative is excimer laser phototherapeutic keratectomy.
- Advanced cases will require keratoplasty: deep anterior lamellar keratoplasty or penetrating keratoplasty. The recurrence rate in the corneal graft is significant and may necessitate repeat surgery. Keratoplasty combined with limbal stem cell transplantation has also been advocated to reduce the recurrence rate.
Epithelial-stromal TGFB1 dystrophies:2
|
Reis-Bucklers corneal dystrophy |
Thiel-Bienke corneal dystrophy |
Lattice corneal dystrophy Type 1 |
|
|
Eponyms |
Corneal dystrophy of Bowman layer type 1 (CDB1), Atypical granular corneal dystrophy, Superficial Granular Corneal Dystrophy |
Corneal dystrophy of Bowman layer Type II; Honeycomb-shaped corneal dystrophy: Anterior limiting membrane dystrophy, type II; Curly fibers corneal dystrophy; Waardenburg–Jonkers corneal dystrophy |
LCD, type 1; Biber-Haab-Dimmer. |
|
Inheritance |
Autosomal dominant |
Autosomal dominant |
Autosomal dominant |
|
Genetic locus |
5q31 |
5q31 |
5q31 |
|
Gene |
Transforming growth factor beta-induced—TGFBI |
Transforming growth factor beta-induced—TGFBI |
Transforming growth factor beta-induced—TGFBI |
|
Onset & course |
Childhood Slowly progressive |
Childhood Slowly progressive |
1st to 2nd decade Progressive with marked reduction in vision by 4th decade |
|
Symptoms |
Recurrent corneal erosions and reduced vision from childhood |
Initially, there are recurrent corneal erosions followed later by the reduction in vision at which time erosions lessen |
Ocular discomfort and pain from recurrent corneal erosions and visual impairment as a result of this in early childhood. Marked visual loss by 4th decade. |
|
Signs |
Confluent irregular whitish-gray geographic opacities (Figure 3 A& B) with varying densities at the level of Bowman’s layer and with time these opacities extend into superficial then deep stroma and also to the limbus. In advance cases the opacities are denser and looks similar to curdled milk appearance (Figure 3 C& D). There may be clear spaces within the irregular opacities. |
Initial lesions are solitary flecks or irregular grayish white scattered opacities at the level of Bowman’s layer. Later there are honeycomb subepithelial opacities that can progress deeper into the stroma. Initially periphery is clear but later periphery is affected. TBCD and RBCD can often be difficult to differentiate clinically in the early stages. Optical coherence tomography can be helpful as it shows hyperreflective opacities at the Bowman’s layer that extend into the epithelium in a typical sawtooth pattern. |
Superficial central fleck like opacities. Initially isolated fine refractile lattice lines. Sub-epithelial white opacities. All the above can be present as early as end of first decade. With progression lines become prominent and extend to the periphery and into the deeper stroma (Figure 4). There is central subepithelial ground glass haze of central and paracentral cornea. Recurrent erosions may be complicated by microbial keratitis. Vision is reduced by the 2nd to 4th decade. |
|
Pathology |
Bowman’s layer is replaced by a sheet-like granular layer that stains red with Masson-Trichrome. This extends into the stroma depending on severity. |
The varying thickness of the epithelium is present due to a thickened abnormal subepithelial fibrous layer that replaces the Bowman layer and has a characteristic sawtooth-like surface. Curly collagen fibers are seen in transmission electron microscopy and immunohistochemistry |
Epithelial layer atrophy. Deposition of eosinophilic amyloid material that stains with Congo red and displays bi-refringence under polarized light (Figure 4D). |
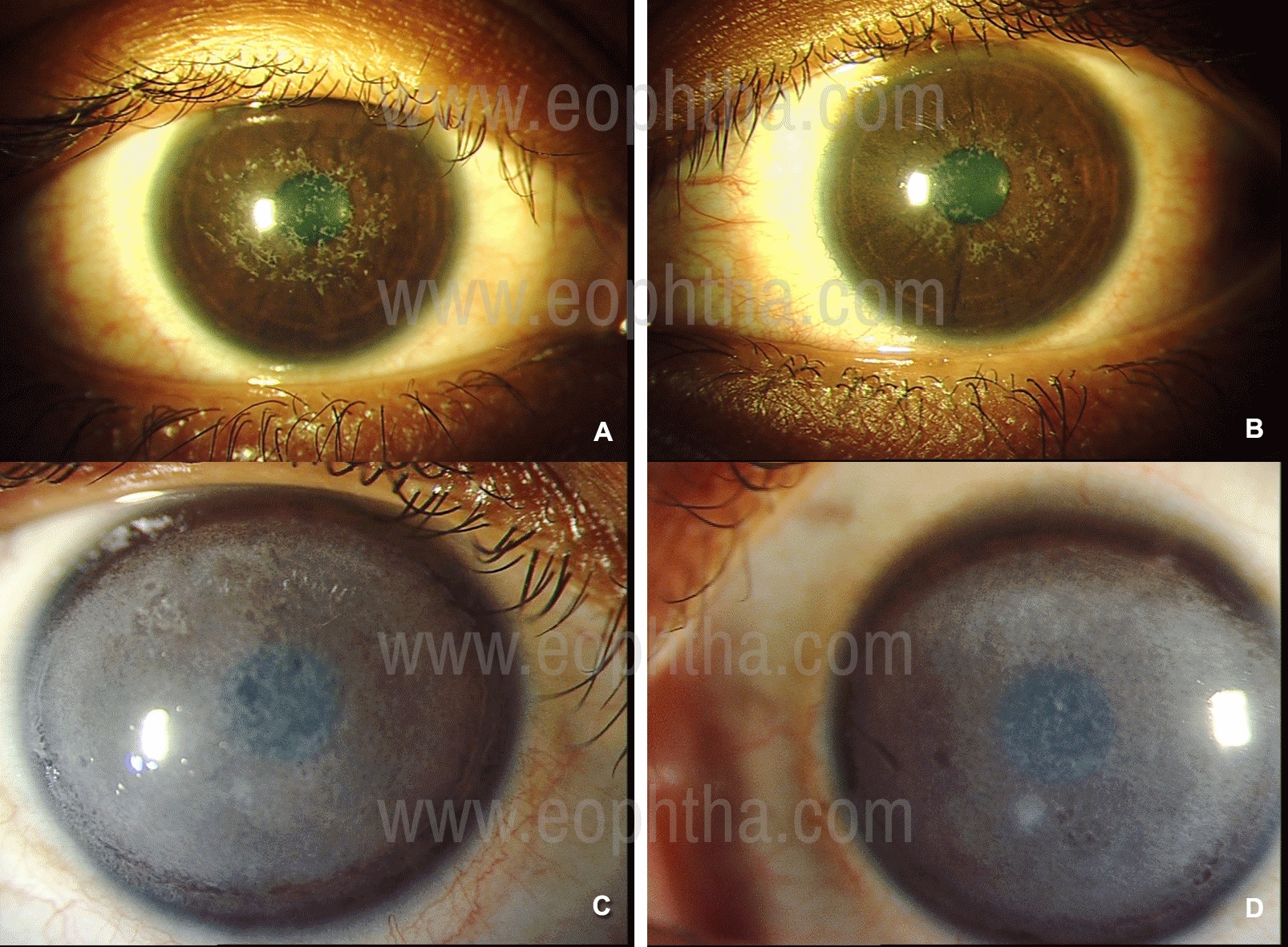
Figure 3: Slit-lamp photographs of Reis-Buckler’s Corneal Dystrophy in a 24-year old female. In direct illumination (A & B) irregular, confluent white opacities are seen in the anterior cornea. When these deposits are dense they appear like curdled milk as seen in another patient, a 17-year old girl who required penetrating keratoplasty in both the eyes (C & D).
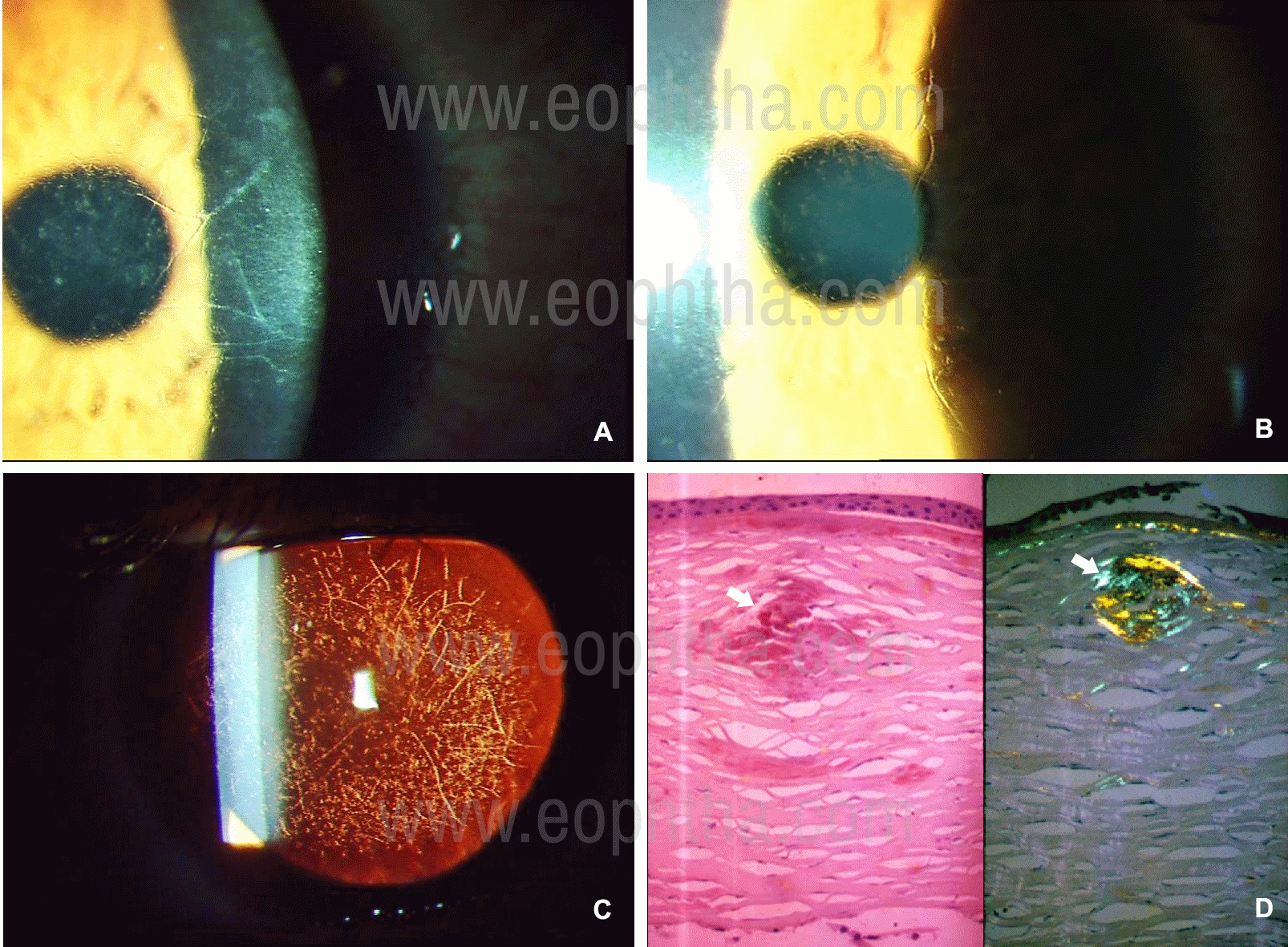
Figure 4: Slit-lamp photographs of Lattice Corneal Dystrophy. disease in a 41-year old lady, whose brother also is affected. Anterior stromal dots, and refractile lines are seen in more clearly in indirect illumination slit-lamp techniques like iris retro-illumination and proximal (A & B). In fundus retro-illumination (C) the dense reticular pattern of lattice lines and dots can be seen. Histology of the cornea (D) stained with Congo red (in the left) shows amyloid deposits (arrow) and the same section viewed in polarized light reveals the deposits to be birefringence and red-green dichroic. (Histology photomicrograph courtesy of Dr J Biswas, MS; Ophthalmic Pathology, Sankara Netralaya, Chennai, India).
It is to be noted that historically Lattice corneal dystrophies have been described with multiple variants.2 This has been mainly due to variations in phenotypic and genotypic presentations. There are more than 2 dozen distinct heterozygous amyloidogenic mutations which are all located to the TGFBI gene. The previously describe lattice corneal dystrophy Type 2 is not corneal dystrophy but familial amyloidosis, Finnish type or gelsolin type which is also known as Meretoja Syndrome. The structure of lattice lines is dependent on age and mutation. Variants like Type IIIA have larger ropy lattice lines extending from limbus to limbus or in Type I/III is thinner or sometimes the lines may be absent. Recurrent erosions are absent in Type IV (small lattice lines) or in those where the lines are absent (polymorphic amyloidosis). Classical Lattice corneal dystrophy is more widespread while the variants are restricted to limited geographic locations, and none of these locations are in India.
Treatment:6
- Asymptomatic patients require no treatment.
- For patients with foreign body sensation, ocular discomfort or recurrent erosions, treatment is similar to that described in the preceding section.
- If vision is significantly reduced then deep anterior lamellar keratoplasty or penetrating keratoplasty is required. There is possibility of recurrence of lesions in the graft.
Epithelial-stromal TGFB1 dystrophies & Stromal dystrophies:2
|
Granular corneal dystrophy (classic) |
Granular corneal dystrophy Type 2 |
Macular Corneal dystrophy |
|
|
Eponyms |
Corneal dystrophy Groenouw type I |
Avellino dystrophy Combined granular-lattice dystrophy |
Groenouw corneal dystrophy type II |
|
Inheritance |
Autosomal dominant |
Autosomal dominant |
Autosomal recessive |
|
Genetic locus |
5q31 |
5q31 |
16q22 |
|
Gene |
Transforming growth factor beta-induced—TGFBI |
Transforming growth factor beta-induced—TGFBI |
Carbohydrate sulfotransferase 6 gene—CHST6. |
|
Onset & course |
Childhood (as early as 2 years) Very slow progression |
Early Childhood Slowly progressive |
Childhood Progressive with fall in vision by 2nd - 3rd decade |
|
Symptoms |
Glare and photophobia. Visual acuity is affected late in disease course. Sometimes epithelial erosions occur |
Vision is affected only late when opacities affect the visual axis. Sometimes epithelial erosions occur |
Profound visual impairment in late childhood and early adult life. |
|
Signs |
Well defined white discrete irregular granular opacities (snow-flake appearance) with clear intervening stroma (Figure 5 B,D,F,H). The periphery is spared. With age, opacities may affect posterior stroma |
Well defined white granular opacities like bread-crumb with star-shaped peripheries, central clear areas within the white opacities. There are lattice lines, which are short, dash-like, less refractile and do not intersect each other (Figure 6). |
Initially white discrete irregular fleck like opacities appear which progresses to involve all layers of the cornea and extend to the limbus (Figure 5 A, C, E, G). The intervening cornea between the opacities has a grayish haze. The cornea is thin. Erosions can occur. |
|
Pathology |
Hyaline opacities are present in the stroma that stain red with Masson’s trichrome (Figure 5H) |
Deposition of both hyaline and amyloid material in the corneal stroma that stain with Masson trichrome and Congo red |
Glycosaminoglycan deposits that stain blue with Alcian blue, that involve the endothelium (Fig 5G). |
NB: Granular dystrophy is included in this section as classically it has always been considered as a stromal dystrophy
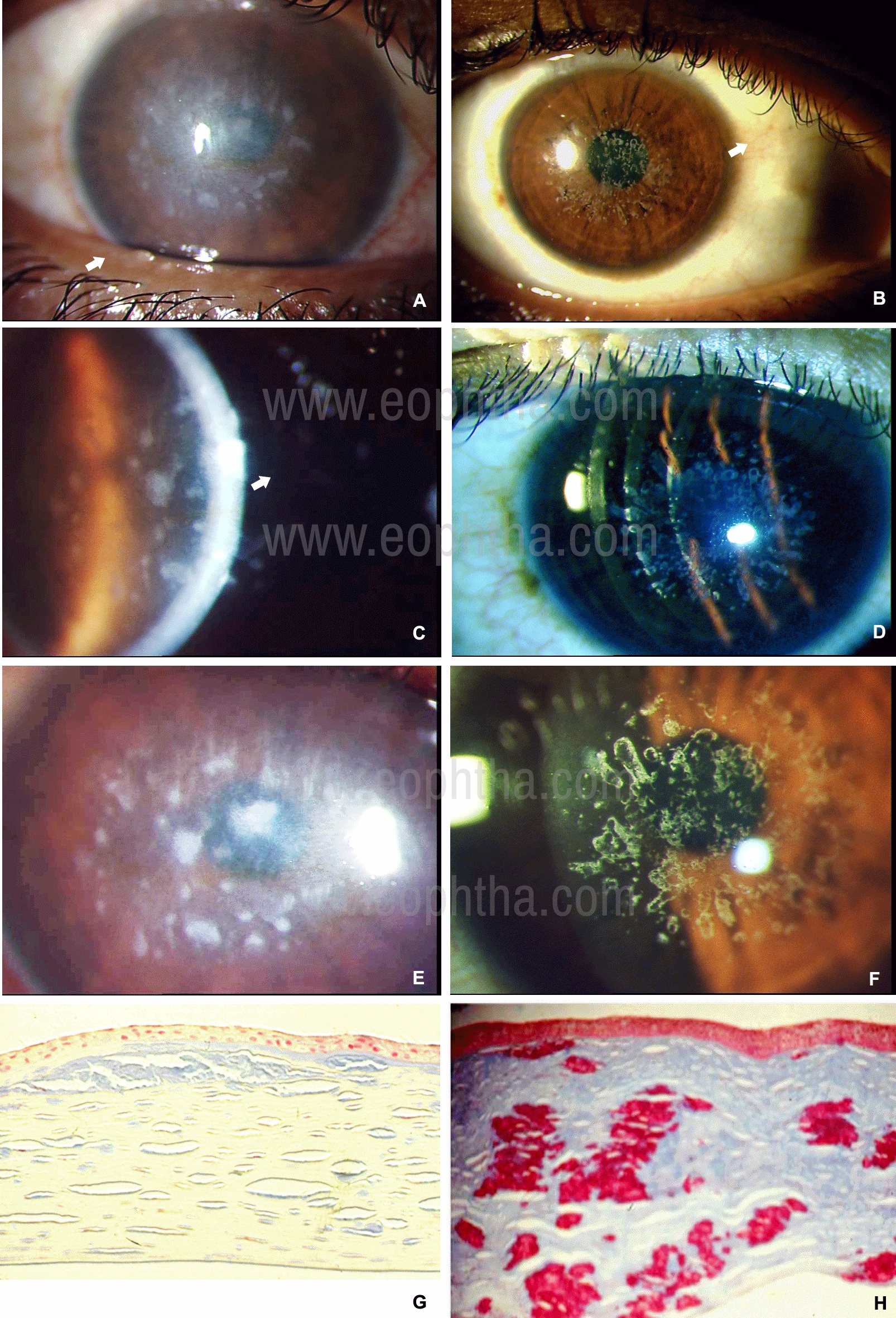
Figure 5: Figure 5: Slit-lamp photographs of Macular Corneal Dystrophy (A,C,E,G) and Granular Corneal Dystrophy (B,D,F,H). In diffuse illumination (A) the whitish-gray deposits of macular corneal dystrophy are irregular with indistinct margins and extending to all layers of the cornea (C). In contrast, the deposits in granular corneal dystrophy are whiter, bread-crumb like with distinct margins (A). Initially, the deposits are limited to the anterior stromal layers (D). Note the cornea between the deposits in macular corneal dystrophy is hazy (E), while in contrast, the cornea between the deposits in granular corneal dystrophy is clear (F). Sections of the stroma stained with Alcian blue (G) show glycosaminoglycans (blue) in macular corneal dystrophy. Sections of the stroma stained with Masson’s trichrome stain (H) shows red-stained keratohyalin deposits in granular corneal dystrophy. (Histology photomicrograph courtesy of Dr. J Biswas, MS; Ophthalmic Pathology, Sankara Netralaya, Chennai, India).
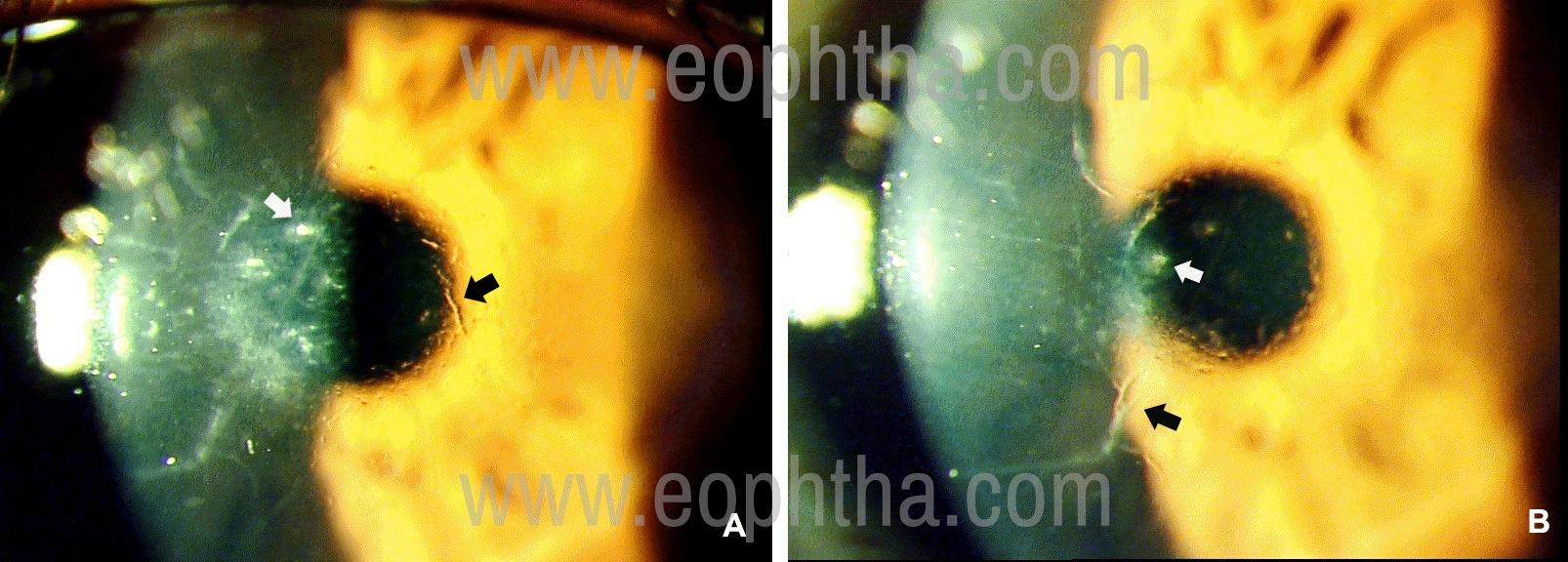
Figure 6: Slit-lamp photographs of Avellino corneal dystrophy (A and B) in a 37-year old male. The right (A) and left (B) eye shows anterior stromal irregular white opacities (white arrows) along with refractile lattice lines (black arrows) which are evident in both direct and indirect slit-lamp illumination.
Treatment:6
- Asymptomatic patients with GCD requires no treatment.
- For epithelial erosions treatment is similar to that described in the previous sections
- Penetrating keratoplasty or deep anterior lamellar keratoplasty is required when visual impairment is significant.
Endothelial dystrophies:2
|
Fuchs endothelial corneal dystrophy |
Congenital hereditary endothelial dystrophy |
Posterior polymorphous corneal dystrophy |
|
|
Eponyms |
- |
- |
- |
|
Inheritance |
Heterogenous and mostly unknown. Some cases have autosomal dominant inheritance |
Autosomal recessive |
Autosomal dominant |
|
Genetic locus |
Multiple |
20p13 |
Multiple |
|
Gene |
Early-onset type: COL8A2 |
Solute carrier family 4, sodium borate transporter, member 11-(SLC4A11) (in majority) |
Multiple |
|
Onset & course |
Commonly in 4th decade or later Progressive |
Congenital Relatively stationary with very slow or no progression |
Childhood Very slowly progression |
|
Symptoms |
Early stages no symptoms may be present and disease is detected on routine ocular examination or following cataract surgery when there is corneal decompensation. In the natural course of the disease there is initially reduced or cloudy vision in the morning on waking up which is transient and clears within few hours. With progression there is delayed clearance and then there is no clearance. This is accompanied by foreign body sensation from epithelial bullae and pain, lacrimation from ruptured bullae. |
Reduced vision and clouding. No photophobia or lacrimation. |
Mostly asymptomatic and detected during ocular examinations. Vision may be affected late if there is corneal edema |
|
Signs |
(Figure 7) Stage 1: Cornea guttate- center to involve the periphery Stage 2: Corneal edema Stage 3: Bullous keratopathy Stage 4: subepithelial fibrosis, scarring and superficial vascularization in longstanding eyes Specular or confocal microscopy: Polymegathism, pleomorphism, decreased hexagonality and reduced number of endothelial cells |
Bilateral often asymmetric clouding of the corneal giving a milky white ground glass appearance, with increased corneal thickness (Figure 8) Intra-ocular pressure is not elevated. |
Asymmetric signs between the two eyes of the same patient. Geographic, vesicular, or linear white parallel lines (rail-track) opacities (Figure 9) on the endothelium. Occasionally corneal edema Steep cornea with high keratometry values |
|
Pathology |
Diffuse thickening of the Descemet’s membrane, with localized hyaline excrescences(guttae) and gradual loss of endothelial cells (Figure 7D) |
Thickened Descemet’s membrane with corneal stromal edema and disruption of the lamellar pattern |
Multi-laminar appearance of the corneal endothelium |
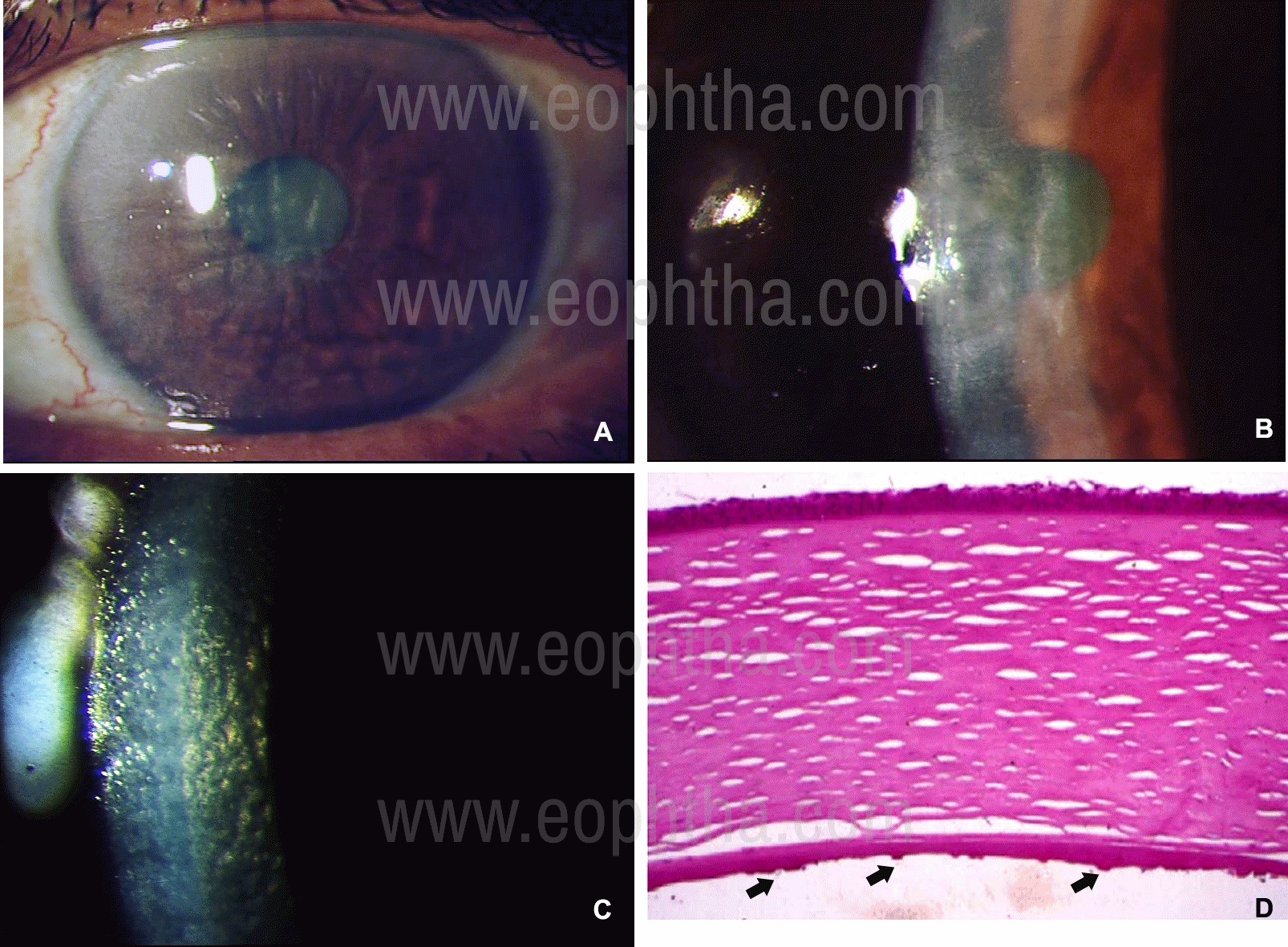
Figure 7: Slit-lamp photographs of Fuchs Endothelial Corneal Dystrophy in a 68-year old lady. There is frank corneal edema with folds in the Descemet’s membrane in the left eye(A & B). Corneal guttate and a beaten metal appearance is seen in the parallelepiped section of the slit-lamp illumination (C). Histology of the cornea reveals hypertrophied epithelial layers, separated corneal lamellae in the stroma and thickened Descemet’s membrane, with complete loss of endothelium and presence of wart-like thickenings (arrows). (Hematoxylin-Eosin stain; Histology photomicrograph courtesy of Dr J Biswas, MS; Ophthalmic Pathology, Sankara Netralaya, Chennai, India).
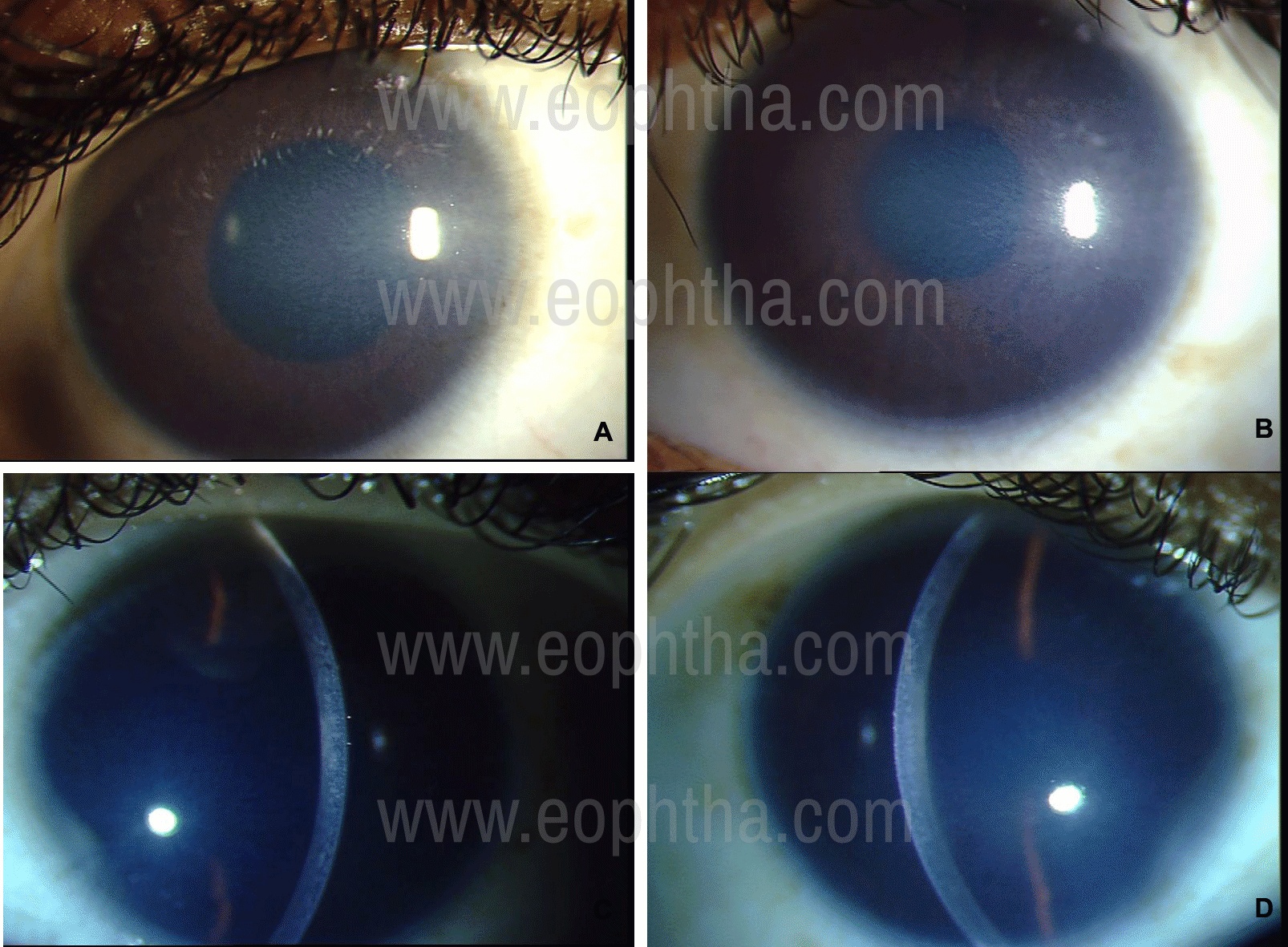
Figure 8: Slit-lamp photographs of Congenital Hereditary Endothelial Dystrophy in a 14-year old girl. Her brother also is affected. There is diffuse ground-glass opacification of the cornea (A & B) in diffuse illumination. The cornea is uniformly thickened as seen in the optic section of the slit-beam (C & D). Her vision at the presentation in 2005 was 20/64 therefore penetrating keratoplasty was deferred. However, her vision slowly deteriorated and she underwent penetrating keratoplasty in both the eyes in 2015.
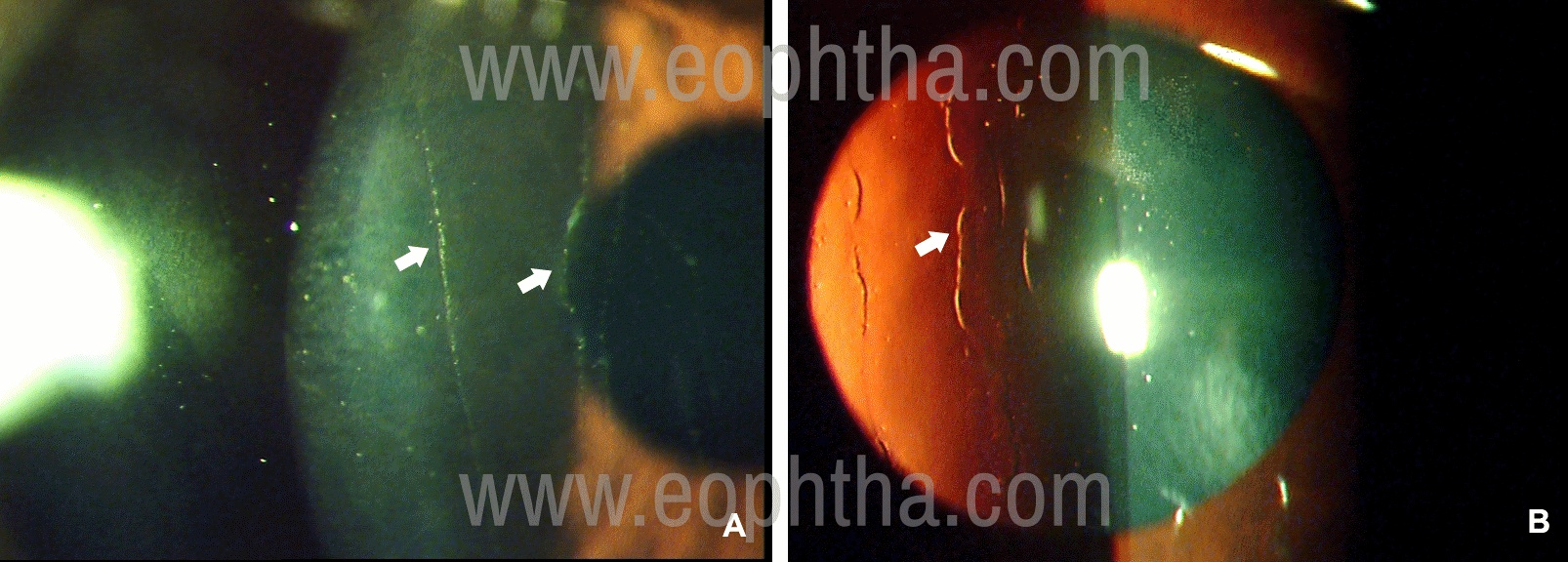
Figure 9: Slit-lamp photographs of Posterior Polymorphous Corneal Dystrophy in a 13-year old girl who had come for a routine eye examination. In direct illumination (A) vertical linear lines (arrows) are seen on the endothelium which are railway-track like in fundus retro-illumination (B).
Treatment:
Fuchs endothelial corneal dystrophy6
- The definitive treatment for Fuchs endothelial corneal dystrophy is endothelial keratoplasty: Descemet’s stripping endothelial keratoplasty (DSEK/DSAEK) or Descemet membrane Endothelial keratoplasty (DMEK). Earlier penetrating keratoplasty was the standard practice but with the advent of endothelial keratoplasty, DSAEK or DMEK is the new standard of care. This is combined with cataract extraction.
- In the presence of bullous keratopathy, topical viscous preservative free lubricants are advised. The role of topical 5% sodium chloride eye drops is questionable. Bandage contact lens can also be advised but these carry the risk of secondary infections. Anterior stromal puncture leads to subepithelial fibrosis and reduction in bullae and is a good palliative treatment. This is advised for localized bullous keratopathy, or in eyes where endothelial or penetrating keratoplasty is not planned. It is better to avoid performing anterior stromal puncture in the pupillary area If future endothelial keratoplasty is considered.
- Newer advances in treatment of Fuchs endothelial corneal dystrophy: Important advances in regenerative medicine in recent times indicate that in future the treatment may be less invasive.9 Cultured endothelial cells and intra-cameral injection of these cultured cells leading to endothelial regeneration is one such method. Descemetorhexis, whereby the central disease endothelium is removed and peripheral endothelial cells repopulate the central cornea is another. The use of topical rho-associated kinase inhibitors to promote endothelial cell proliferation either alone or in conjunction with descemetorhexis or cultured endothelial cell transplantations has been reported in small case series to be also beneficial.
Other considerations and Cornea guttate
- Cornea guttate are often detected during routine ocular examination and commoner in females. These necessary do not progress to Fuchs endothelial corneal dystrophy. However, these patients may be monitored and a baseline specular microscopy and corneal pachymetry can be advised for more precise follow-up. The patients should be explained about the condition without causing undue alarm.
- If corneal guttate are detected when the patient has presented for cataract surgery then specular microscopy and corneal pachymetry is required particularly the latter for better evaluation of corneal endothelial function. A history of morning blurring of vision that clears up should alert the ophthalmologist of deteriorating endothelial function in presence of hypoxic stress.
- Routine phacoemulsification can be performed using low energy and fluidics, Viscoat® viscoelastic device and balance salt solution. Care should be taken to avoid endothelial damage during surgery. If the cataract is hard then small incision or extra-capsular cataract surgery should be performed depending on the surgeon’s expertise. It is preferable that the surgery in these patients is performed by experienced surgeons. Patients should be cautioned about the development of corneal edema after cataract surgery in their lifetime and should be periodically examined lifelong.
- The presence of cataract in a patient with Fuchs endothelial corneal dystrophy often leads to management dilemma whether to perform only cataract surgery or to combine it with endothelial keratoplasty (triple procedure). Several criteria like central cornea thickness10 and parameters from Scheimpflug tomography11 have been published to guide surgeons in decision making. If there is no sign of corneal edema it is advisable to perform only cataract surgery. It would be prudent to explain to the patients that endothelial keratoplasty still might be required in the near future.
Posterior polymorphous corneal dystrophy6
- Treatment principles are similar to the above section.
- The definitive treatment is DSEK/DSAEK or DMEK.
Congenital Hereditary Endothelial Disease6
- Penetrating keratoplasty or endothelial keratoplasty
- Surgery should be performed early to avoid amblyopia
Corneal dystrophies in India
There are no published reports on the frequency or pattern of corneal dystrophies in India. There is one report that gives the pattern of corneal dystrophies that required corneal transplantation in a tertiary referral center in South India (see the box below).12 The pattern is different from the west, where Fuchs endothelial corneal dystrophy is more commonly reported.
|
Corneal dystrophy |
Number (percentage) |
|
Congenital hereditary endothelial dystrophy |
63(34.8) |
|
Macular corneal dystrophy |
53(29.3) |
|
Fuchs endothelial corneal dystrophy |
30(16.6) |
|
Lattice corneal dystrophy |
15(8.3) |
|
Gelatinous drop-like corneal dystrophy |
8(4.4) |
|
Posterior polymorphous corneal dystrophy |
5(2) |
|
Granular corneal dystrophy |
4(2.2) |
|
Reis-Buckler’s corneal dystrophy |
3(1.7) |
Table 2. The pattern of Corneal dystrophies undergoing penetrating keratoplasty in India12
Differential diagnoses of corneal dystrophies
There is a wide variation in the clinical presentation of corneal dystrophies due to phenotypic heterogeneity. This is due to the expressivity of the genetic mutation.3 While there may be confusion on the clinical presentation between different corneal dystrophies, other corneal lesions may also confuse the clinician.
The primary requisite for making a correct diagnosis is familiarity with the clinical features of all the corneal dystrophies. It is not possible to remember all the clinical signs, therefore keeping an atlas of clinical signs in the clinic is helpful. Secondly, one should be adept at the use of the slit-lamp and use the various illumination techniques, particularly indirect illumination techniques, routinely. The slit-beam should be swung from one side of the cornea to the other, and the cornea scanned from side to side. Non-invasive imaging techniques like confocal microscopy or optical coherence tomography can provide additional help. The final diagnosis is by histology.
The following points are always helpful in arriving at diagnosis: 6
- The symmetry of the corneal lesions in both the eyes. Corneal dystrophies are usually symmetrical.
- Rule out signs of inflammation (including past) like vascularization, ghost vessels, scleral changes, keratic precipitates, history of redness and pain. The presence of these signs indicate that the corneal lesions may not be a dystrophy
- Localize the opacities to the corneal layers: epithelial, sub-epithelial, stromal, pre-Descemet or endothelial
- Is there a clear cornea between the opacities? This distinguishes granular from macular dystrophy.
- The thickness of the cornea: normal, thick, or thin.
- The presence of family members with similar symptoms or signs (positive family history) rules in favor of dystrophy.
|
Diseases |
Corneal dystrophy |
Differentiating features |
|
Bilateral corneal scars |
Any corneal stromal dystrophy |
Asymmetrical, variation in shape, associated vascularization, history of infection/inflammation |
|
Spheroidal degeneration |
Gelatinous drop-like corneal dystrophy |
Asymmetrical presentation, discrete or sometimes confluent brown spheroidal lesions which are predominantly in the nasal & temporal periphery in the interpalpebral region, older age group, vascularization |
|
Post-traumatic corneal erosions |
Epithelial & subepithelial dystrophies |
H/o trauma, unilateral, loosely adhered epithelium which is localized |
|
Corneal keloid |
Gelatinous drop-like corneal dystrophy |
Unilateral in acquired. Bilateral in congenital corneal keloid – will have associated systemic or other ocular anomalies |
|
Vertical rupture of Descemet’s membrane |
Posterior polymorphous corneal dystrophy |
H/o birth trauma, vertical in orientation, unilateral or bilateral asymmetric |
|
Cornea farinata |
Pre-Descemet’s corneal dystrophy |
Dot or comma like opacities in the deep stroma adjacent to the endothelium. Faint opacities that are best visible in retro-illumination. Anterior stroma is normal and vision is not affected. Can be bilateral. Confocal microscopy is helpful |
|
Polymorphic amyloid degeneration |
Lattice corneal dystrophy |
Appears in older persons. Opacities are stellate flecks with irregular refractile filaments. Vision is normal |
|
Salzmann nodular degeneration |
Gelatinous drop-like corneal dystrophy |
Grayish nodules arranged in a circle in the peripheral or paracentral cornea. Usually associated with chronic keratitis. Vascularization is usually present along with a pannus. |
|
Hassall-Henle bodies |
Fuchs endothelial corneal dystrophy |
Appears like corneal guttate but is present in the corneal periphery and is present in elderly individuals |
|
Vortex keratopathy (cornea verticillate) |
Epithelial dystrophies |
Unilateral or bilateral. Golden-brown or gray superficial deposits seen in inferior cornea arranged in a clock-wise whorl pattern. History or drug intake like amiodarone, chloroquine, etc. |
|
Inherited metabolic disorders – Mucopolysaccharidosis syndromes (Hurler’s syndrome, Schie’s syndrome, Maroteaux-Lamy syndrome, Morquio syndrome); Mucolipodosis, Lysosomal storage disease, Hypolipoproteinemias, miscellaneous metabolic diseases |
Congenital hereditary endothelial dystrophy |
Clouding of the cornea is present in infants or young children and sometimes in older children (Morquio syndrome). They are associated with other ocular or systemic anomalies. In some of these there is crystalline deposition in the corneal stroma |
|
Primary systemic amyloidosis (Familial amyloidosis) |
Lattice corneal dystrophy |
Lattice lines are less numerous and central cornea is spared. Corneal sensation is reduced. The characteristic mask like facies, cranial and other nerve palsies, dry lax skin are other clues to the clinical diagnosis |
|
Iridocorneal endothelial syndrome |
Corneal endothelial dystrophies |
Unilateral. Prominent iris and pupillary abnormalities. |
|
Congenital glaucoma |
Congenital hereditary endothelial dystrophy |
Increased corneal diameter. Corneal edema is not uniform across the cornea and the corneal thickness is not increased. Raised intra-ocular pressure |
Table 3. Differential diagnoses
|
Autosomal recessive |
|
|
X-linked |
|
|
Unknown |
|
|
Autosomal dominant |
|
Table 4. Summary of the mode of inheritance of corneal dystrophies2
|
Clinical presentation |
Corneal dystrophies |
|
Early childhood with visual impairment |
|
|
Early childhood but no visual impairment |
|
|
First to Second decade with progressive visual impairment |
|
|
First to Second decade with little or no visual impairment |
|
|
Third to Fourth decade or later with visual impairment |
|
Table 5. Corneal dystrophies according to age and visual impairment13
References:
1. Weiss JS, Moller HU, Aldave AJ, et al. The IC3D classification of the corneal dystrophies. Cornea 2008;27(Suppl.2):S1-S42. 2. Weiss JS, Moller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies - Edition 2. Cornea 2015;34:117-59. 3. Aldave AJ. The genetics of corneal dystrophies. Dev Ophthalmol 2011;48:51-66. 4. Waring GO, Rodrigues MM, Laibson PR. CorneaI Dystrophies. I. Dystrophies of the epithelium, layer and stroma. Surv Ophthalmol 1978;23:71-122. 5. Waring GO, Rodrigues MM, Laibson PR. CorneaI Dystrophies. II. Dystrophies of the endothelium. Surv Ophthalmol 1978;23:147-68. 6. Corneal dystrophies and ectasias. In External diseases and Cornea. Basic and Clinical Sciences 2019-2020. Eds: Weisenthal RW, Daly MK, de Freitas D, et al. American Academy of Ophthalmology San Francisco, CA.pp:166-216. 7. Reidy JJ, Paulus PM, Gona S. Recurrent erosion of the cornea. Epidemiology and treatment. Cornea 2000;19:767-71. 8. Kaza H, Barik MR, Reddy MM, et al. Gelatinous drop-like corneal dystrophy: a review. Br J Ophthalmol 2017;101:10-15. 9. Sarnicola C, Farooq AV, Colby K. Fuchs endothelial corneal dystrophy: update on pathogenesis and future directions. Eye & Contact Lens 2019;45:1-10. 10. Van Cleynenbreugel H, Remejier L, Hillenar T. Cataract surgery in patients with Fuchs’ endothelial corneal dystrophy. When to Consider a Triple Procedure? Ophthalmology 2014;121:445-453. 11. Patel SV, Hodge DO, Treichel EJ, et al. Predicting the prognosis of Fuchs endothelial corneal dystrophy by using scheimpflug tomography. Ophthalmology 2019;127:315-23. 12. Pandrowala H, Bansal A, Vemuganti GK, Rao GN. Frequency, distribution, and outcome of keratoplasty for corneal dystrophies at a tertiary eye care center in south India. Cornea 2004;23:541-546. 13. Klintworth GK. Corneal dystrophies. Orphanet Journal of Rare Diseases 2009;4:7. doi:10.1186/1750-1172-4-7.