
Twenty first century has seen a giant leap in the understanding of human genetics. Based on the human genome project humans are supposed to have roughly 20,000 to 25,000 genes. Genes are the fundamental units of heredity. To understand genetics involved in various ocular pathologies a basic understanding is essential. The following discussion will provide an overview of basic concepts in genetics and provide a glimpse into the genetics of Retinoblastoma and intraocular melanoma.
The basic structure of a chromosome[1]
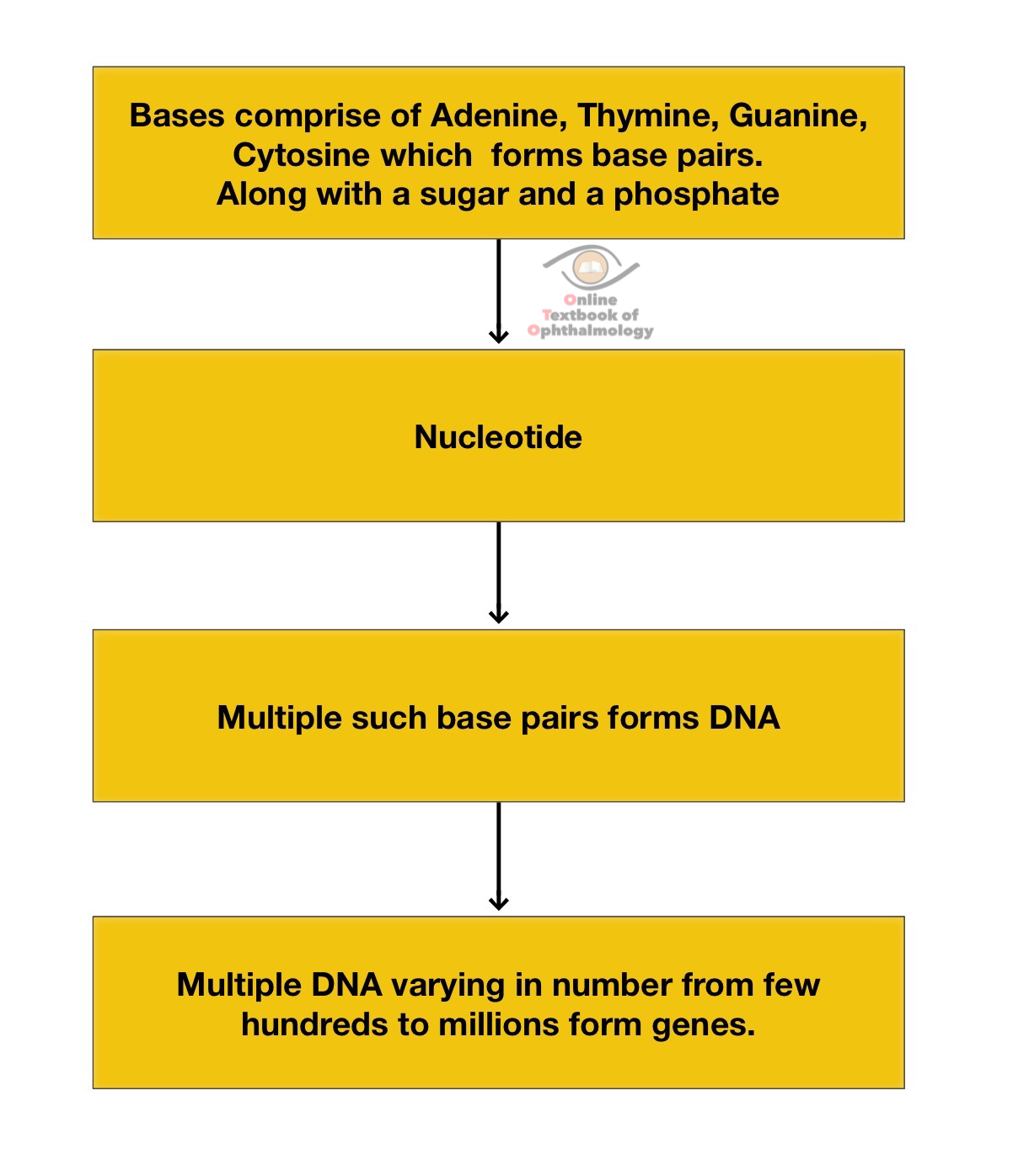
- Chromosomes are formed by a tight packaging of the DNA stored in the nucleus of a cell
- Each gene in humans are comprised of 2 copies ; one inherited from either parent.
- Forms of the same gene with differences on the basis of bases are known asalleles.
- A tightly regulated process oftranscriptionandtranslationinvolves the production of basic building blocks called as amino acids to form proteins.
- Gene regulation is the process whereby certain genes are'switched on'and others are silenced.
- The process of cell replication and division is a highly regulated process wherein each and every step is checked ; any error is corrected when possible, if not the cell undergoes death by a process called asapoptosis.
- Each chromosome has as short(p) and long arm(q) joined at thecentromere.
Taking the example of retinoblastoma gene lets see how a chromosome is named.
- the first number before a alphabet denotes the chromosome number.
- the alphabet 'p' or 'q' symbolizes the small or long arm of a chromosome.
- the number after the alphabet is suggestive of the band position on a chromosome.
- Abbreviation like 'cen' indicates location close to centromere, 'ter' indicates the location close to the terminal part of a choromosome
- So 13q14 would indicate the location to be in the long arm of chromosome 13, near band 14.
MUTATION[1]
Any structural derangement in a gene is termed as mutation. Mutations can be
- Hereditary : In this the mutations are inherited from either of parent(who may or may not be affected) with all the cells of the index patient having the concerned mutation. Since the parent's egg or the sperm( germ cells) might have had the same mutation, it is also known as germline mutation. The index case has the chances of passing ( hereditary) it to his offspring's.
- Somatic: The index case has the concerned mutation in later part of its life and the concerned DNA changes are seen in the somatic cells( sparing the germ cells). There are no chances of passing it further.
- De novo mutations can be hereditary or somatic. In some cases the mutations occur only in the germ cells of the parent (without them being affected) which gets passed on. In another scenario the mutation occurs just after fertilization. In both such forms the affected person has the mutation afflicting all his cells and has the chances of passing it on. Interestingly even if the person has the chances of passing the mutation further on , none of his parents would have been affected as the mutation is 'de novo'.
Types of Mutation
Each amino acid is coded by group of 3 bases
- Missence mutation: replacement of a base pair by another: leads to a different amino acid.
- Nonsense mutation: a replacement of a base pair by another leading to premature stoppage in the process of coding a protein thereby forming a truncated protein.
- Insertion: addition of additional bases
- Deletion: removal of base pair without any replacement
- Duplication
- Frame shift mutation: addition or deletion of base pairs leading to altered gene's reading frame . This leads to alterations in the grouping of the base pairs forming different amino acids.
- Repeat expansion: Repetition of short DNA sequences in a row.
Structural chromosomal changes
Translocation:when a piece of chromosome breaks and attaches to another.
Deletion:when a part of chromosome gets lost.
Duplication
Inversion:when chromosome breaks at two places and the resultant pieces gets re- inserted into opposite ends
Isochromosomes:In place of a short and long halves, the chromosome have either 2 long or 2 short halves
Dicentric chromosomes:Presence of 2 centromeres secondary to abnormal fusion.
Ring chromosomes:when chromosome breaks at two places with the resulting ends joining each other.
Degrees of Relationship
|
Degrees of relationship |
Examples |
|
First degree |
Parents,Children,Brothers and sisters |
|
Second degree |
Grandparents, grandchildren,uncles and aunts,nephew and nieces |
|
Third Degree |
Cousins |
Inheritance Patterns[1]
Autosomal dominant
- Only one copy of the defective gene is required to exhibit the disease.
- Persons affected have 50% chances of passing it on to future generations.
- Unaffected persons don't have any risk of passing it on.
- Each generation gets affected
- Males and females are equally afflicted
Autosomal Recessive
- Affected patients have 2 copies of the defective gene to exhibit the condition.
- Affected persons have unaffected parents each carrying a single copy of the defective gene.
- 25% chances of offspring having the condition when parents have the defect.
- Not all generations gets involved.
X linked dominant
- Mutation as a result of changes in the X chromosome
- The inheritance pattern depends on whether the father or mother is affected.
- If the father is affected only his daughters exhibit the condition.
- No male to male transmission
- Females are more affected than males.
X linked recessive
- Males gets affected more
- Females are generally the carriers
- No male to male transmission
Codominant
Mitochondrial
Genetics in Retinoblastoma[4],[5]
Retinoblastoma(RB) is one of the very few malignant ocular conditions, where having a sound knowledge of genetics provides treatment just beyond eye care. It was the Retinoblastoma gene(RB); which was one of the earliest tumour suppressor gene to be mapped and studied which then opened the door to where we are in the current understanding of cancer genetics. The brilliant paper by Knudson[2],[3] whose theory about the 'double hit' i.e a malignant change can be observed when at least 2 'hits' or mutations take place revolutionized the science behind understanding malignancies. The RB gene has been mapped to 13q14.RB gene functions as a tumor suppressor. Any damaged DNA is prevented from replicating further by halting the progress of cell cycle between G1 and S phase. In the following few paragraphs the basics of retinoblastoma genetics would be dealt. From a genetic viewpoint 3 forms of RB can be seen
Familial Retinoblastoma
- Incidence of 10%
- the inheritance of the RB1 gene mutation is from either of the parents who harbour the concerned mutation.
- every cell in the body of the index case has RB1 gene mutation(first hit) followed by a second hit in the retinal cell after conception
- generally bilateral and presents at an early age
Sporadic heritable Retinoblastoma
- Incidence of 30%
- No positive family history
- the first hit develops 'de novo' in the germline of either of the parents (mostly paternal)
- the second hit occurs sometime either just after conception or later.
- the chances of passing on the mutation in consecutive generations is like in a familial retinoblastoma scenario.
- can be bilateral or unilateral but multifocal
Non-heritable Retinoblastoma
- Incidence of 60%
- Both hits of mutation occurs in somatic cells after conception
- no risk to future generations
- unilateral
Few salient points
- All bilateral cases are familial, the reverse is not true
- All unilateral cases may or may not be non heritable. In 10-15%, familial cases may have unilateral presentation.
- the mode of transmission of the RB1 gene mutation is dominant (refer to previous discussion)
- the penetration of RB1 mutation is high (90%).
- in rare cases the penetrance is low with resulting Retinoma.
- Genetic counselling is a complex although rewarding session whereby risk of developing RB in future generations or the current generations are mapped.
- "Every child with RB should undergo tests to rule out a mutation before labelling a case non heritable"
Genetics in Uveal Melanoma [6], [7]
Choroidal melanoma is the commonest ocular malignancy in adults generally occurring in the sixth decade.
The role of genetics in Uveal melanoma is a comparatively newer territory; although a very important one. Various new insights have proven the role of genetics in prognosticating a case of uveal melanoma which is an important asset not only to the physician but also for the patient.
Salient Points
- Chromosome 3: this is the most common chromosomal anomaly reported in Uveal melanoma. Loss of one or two copies of chromosome 3 (monosomy 3) is generally associated with a poorer prognosis in terms of metastasis and survival
- Chromosome 8: gain of chromosome 8 is another poor prognostic criteria.
- Monosomy 3 with a gain of chromosome 8 has the worst prognosis in terms of metastasis and survival.
- Other chromosomal anomalies include, loss of chromosome 1 and 6, however gain of chromosome 6 signifies a better prognosis
- Gene expression profiling(GEP) which measures the activity of many genes divides the uveal melanoma into 2 classes.
- Class 1: has low metastatic potential
- Class 2: has higher metastatic potential
- Although many methods are available for genetic testing, MLPA( multiplex ligation-dependent probe amplification, PCR based) is one of the most sensitive to detect such changes.
- Mutation in other tumour suppressor genes like the BAP1 , increases the chances of multiple malignancies in a patient of uveal melanoma.
References
1) Help Me Understand Genetics. Genetics Home Reference - http://ghr.nlm.nih.gov. Published November 2, 2015.
2) KnudsonAG. Hereditary cancer: two hitsrevisited. J Cancer Res Clin Oncol.1996;122(3):135-40
3) Gaikwad N,Vanniarajan A,Husain A,Jeyaram I,Thirumalairaj K et al. Knudson's hypothesis revisited inIndianretinoblastomapatients. Asia Pac J Clin Oncol.2015 Dec;11(4):299-30
4) Singh AD, Damato BE, Pe'er J, Murphee AL, Perry JD. Clinical Ophthalmic Oncology .Philadelphia: Saunders, Elsevier 2007
5) Claude Houdayer, Marion Gauthier-Villars, Laurent Castéra, Laurence Desjardins, François Doz and Dominique Stoppa-Lyonnet (2012). Retinoblastoma - Genetic Counseling and Molecular Diagnosis, Retinoblastoma: An Update on Clinical, Genetic Counseling, Epidemiology and Molecular Tumor Biology, Prof. Govindasamy Kumaramanickavel (Ed.), ISBN: 978-953-51-0435-3, InTech, Available from: http://www.intechopen.com/books/retinoblastoma-an-update-on-clinical-genetic-counseling-epidemiology-andmolecular-tumor-biology/retinoblastoma-genetic-counseling-protocols-and-molecular-diagnostic-protocols.
6) Onken MD,Worley LA,Char DH,Augsburger JJ,Correa ZM. Collaborative Ocular Oncology Group report number 1: prospective validation of a multi-gene prognostic assay in uveal melanoma. Ophthalmology.2012 Aug;119(8):1596-603
7) Kivelä T,KujalaE. Prognostication in eye cancer: the latest tumor, node, metastasis classification and beyond. Eye (Lond).2013 Feb;27(2):243-52.